Abstract
Although much has been published on the domain structures of human alpha-fetoprotein (AFP), the AFP third domain (AFP-3D) has emerged as an important fragment regarding the binding, docking, and interaction sites for hydrophobic ligands, multiple receptors, ion channels, and cell cycle proteins. In keeping with previous reports, studies have shown beyond doubt that certain amino acid (AA) sequences on AFP-3D provide a docking interface for protein-to-protein interactions (complexing) for such proteins. By means of a computer software program designed to study such “in silico” interactions, certain AA sequences on AFP-3D were identified which could plausibly interact with a group of DNA damage-sensing and repair (DDSR) proteins. The DDSR proteins identified included: 1) BRCA1 and BRCA2 2) FANC1 and FANCD2 3) nibrin 4) ATM and ATR and 5) DNA-PK kinase. Following the mapping of the AFP-3D with DDSR protein interaction sites, the computer-derived AFP-AA identification sequences were examined for similarities and comparisons to previously reported ligand, receptor, channel and other protein interaction sites on AFP-3D. Literature searches revealed that the association of AFP with the DDSR proteins showed correlations not only with clinical serum AFP levels, but also with an intracytoplasmic nonsecreted form of AFP, which interacts with transcription factors, cell death (apoptosis) proteins, nuclear receptors, and enzymes (caspases). The DDSR proteins that interacted with AFP were also found to be involved with cell cycle checkpoint proteins, cyclins and their dependent kinases, and ubiquitin ligases. Finally, both the clinical and experimental reports on the AFP-3D association with DDSR proteins were consistent with the “in silico” findings of this report.
Key Words
Alpha-fetoprotein, DNA repair, BRCA proteins, chromosome instability, Fanconi anemia
Introduction
Human alpha-fetoprotein (HAFP) has a long history of clinical use as a tumor-associated biomarker, employed to detect both fetal defects during pregnancy and adult cancers.[1, 2] Moreover, much of the biochemistry of the HAFP polypeptide has been elucidated over the five decades since AFP was first discovered. HAFP is a single chain polypeptide with an average molecular mass of 69 kDa, depending on its carbohydrate micro-heterogeneity.[3, 4] The secondary structure of this oncofetal protein exhibits a triplicate domain molecular structure, configured by intramolecular loops dictated by 15 disulfide bridges culminating in a helical V- or U-shaped structure.[3] This fetal protein has been classified as a member of the albuminoid gene family, consisting of AFP, albumin, alpha-albumin, vitamin D binding protein, and the AFP-related (ARG) protein.[5] Similar to albumin, HAFP binds to a vast array of ligands, including various drugs, dyes, steroid hormones, heavy metals, flavonoids, fatty acids, and phytoestrogens.[6] Unlike albumin, AFP has proven to be a notable growth factor capable of either cellular enhancement or inhibition.[7].
HAFP is known to bind to multiple cell surface receptors and intracytoplasmic proteins. Recent reviews by the author (GJM) and others have reported the existence of at least three major groups of cell surface receptors, namely, 1) including the scavenger receptor protein family 2), the mucin glycoprotein superfamily, and 3) the chemokine receptor family of proteins.[8-10] The intracellular HAFP binding proteins encompass the a) retinoic acid receptor b) the caspases c) PI3K/AKT (protein kinase-A), d) mTOR e) GAAD153 and f) PTEN. [11, 12] During the last decade, the carboxy-terminal third domain of HAFP (AFP-3D) has been confirmed to be a major binding interface for both cell surface receptors and hydrophobic ligands. [13, 14] Furthermore, the AFP-3D has been touted as a promising agent (fragment) for the selective delivery of anti-cancer agents.[15- 17] Recombinant fragments of AFP-3D have been produced which demonstrate high purification yields, good efficiency of expression, recoverable refolding capabilities, and retention of biological activities. [18-20] In some instances, the AFP-3D recombinant fragment behaves similarly to full-length AFP, while maintaining its capabilities to bind cell surface receptors and intracytoplasmic proteins.[19]
The localization of additional protein binding and interaction sites on AFP-3D, other than the three major receptor and intracellular binding sites mentioned above, continues to be a topic of focus in the biomedical literature. The pursuit to identify additional protein binding/interaction sites on AFP-3D fragments has not abated. Activity sites of interest include receptor blockade and/or inactivation, decoy ligand binding, blunting receptor responses, selective delivery of drugs, and nucleotide agents (miRNAs) and other cargos that are transported into cancer cells or other targeted cells. Such participating cells include lymphoid/leukemic cells, monocytes, macrophages, T-cells, dendritic cells, and various bone marrow cells (stem cells). Thus, knowledge gained from such activities of AFP could conceivably make it possible to modulate, control, and monitor target site interactions and might affect, dictate, or influence signal transduction pathways.
Aims and Objectives
The aims of the present review and prospectus were to search out, identify and localize, and describe plausible sites of interaction of DNA damage-sensing and repair (DDSR) proteins on the AFP-3D fragment. To achieve these aims, computer modeling and molecular software were used to pinpoint sites of possible interaction between the AFP-3D fragment and various proteins of the DDSR protein pathways. The identified proteins and their respective AFP-3D amino acid docking sequences are discussed concerning their relevance to protein-to-protein binding interactions and possible outcomes for DNA repair. Computer modeling and analysis were also employed to compare the DNA-repair protein localized sites to the ligands, receptors, and protein interaction sites previously localized on the AFP-3D fragment. Members of the DNA-repair pathways identified by this process are addressed regarding their biological activities with other ligand and protein interaction sites on AFP-3D. Finally, prior experimental and/or clinical reports of AFP-derived peptide interactions with DNA-repair proteins are addressed in view of their present “in silico” localizations.
Computer Molecular Docking Software
The computer modeling and molecular docking interaction sites of the DDSR proteins were identified and localized by use of a proprietary computer software (Peptimer Discovery Platform) developed and generously provided by Serometrix, LLC (Pittsfield/Syracuse, NY). This software tool was described in detail in earlier publications.[8, 21, 22] Use of the software simulation of protein-to-protein interaction site localization has been repeatedly confirmed and validated by means of in vitro cell-based assays and microarray analyses including receptor binding kinetics. Previous experimental verifications of AFP- 3D interaction sites using this software simulation have included cell cycle proteins, scavenger receptors, immunodeficiency-associated proteins, chemokine receptors, selective and non-selective cation channels, and lysophospholipid and mucin receptors.
DNA Damage Sensing and Repair
Most, if not all cancer cells, have an unstable genome comprising DNA-damaged pathways. In fact, it is uncommon to find a single tumor without a genetic defect. Genomic instability arises either from losing telomeres from the end of a chromosome or from breaks in the DNA contained in the chromosome. After a cell has divided multiple times, its telomeres become critically short. Often, the cell either dies or stops growing, as in end-stage differentiation or aging. If the cell does not stop dividing (i.e., cancer), it leaves chromosomes with broken ends and DNA breaks in mid-chromosome regions. Such breaks are meant to be addressed by a DNA repair mechanism to restore the damage, but if neglected or bypassed, can lead to loss of gene function and a predisposition to cancer.
Since DNA damage can lead to cancer, the cell possesses an intrinsic repair response to DNA damage and to agents causing it. Many human cancers are related to mutations that affect proteins involved in a cellular DNA damage response. For example, DDSR protein mutations in ataxia telangiectasia-mutated (ATM) and Fanconi Anemia (FANCD2) genes [23] can be linked directly to a predisposition to both leukemias and lymphomas. Mutations in other DDSR proteins, such as p53, BRCA1, and BRCA2, can cause ovarian and breast cancers. Mutations in others, such as ATM kinase, are the root causes of chromosome instability in DNA repair disorders which lead to lymphoid and leukemic cancers. When nuclear DNA is damaged, cells rely on specific intracellular signaling pathways to halt cell division before the DNA is copied into another cell. Two such pathways are the cell cycle ATM-CHK2 (checkpoint-2) and the ATRCHK1 (checkpoint-1) pathways.
DNA Repair Proteins
1) The BRCA1 and BRCA2 Proteins
The breast cancer susceptibility genes BRCA1 and BRCA2 were the first breast cancer genes to be identified. BRCA1 and BRCA2 display autosomal inheritance, and the primary tumors are associated with female breast and ovarian cancers. Mutations in BRCA1 and BRCA2 proteins occur in 10-30% of women with germline alterations; such alterations inactivate the BRCA2 allele, while a second allele is inactivated by somatic mutations.[24, 25] Both the DDSR genes are known to participate in homologous recombination pathways and cell cycle control.[26] Interestingly, many of the characteristics of the BRCA2 protein are similar to the FANCD1 gene (see below), and BRCA1 proteins share biological effects common to both proteins. The FANCD1/BRCA2 and BRCA1 proteins interact by binding and forming multi-protein complexes with FANCN proteins, and these complexes function in the DNA repair pathways.[27] Moreover, the FANC and BRCA, RAD51, and CHEK2 proteins can work in concert as multi-protein complexes in the repair of DNA damage.
A) BRCA1. The BRCA1 gene is located on the long q-arm of chromosome 17, consisting of 1,863 amino acids, which encompasses four major domains including a 1) zinc finger (C3HC4 type) 2) nuclear localization signal 3) nuclear export signal motif and 4) BRCA1 C-terminus (BRCT) domain. There are six isoforms which are known to be associated with BRCA1. The human gene encodes a tumor suppressor protein that is responsible for repairing damaged DNA and for destroying cells when DNA cannot be repaired. BRCA1 is also involved in the repair of chromosomal damage, with a role in the repair of DNA double-stranded breaks.[28, 29] If BRCA1 is damaged by mutation and DNA damage is not properly repaired, these events may increase the risk for breast cancer. BRCA1 and BRCA2 are known as proto-oncogenes termed “breast cancer susceptibility type 1 genes” and code for proteins regulating cell growth and differentiation in cells of breast and other tissues. BRCA1 can combine with other proteins, such as tumor suppressors, DNA damage sensors, RNA polymerase-II, and histone deactylase to form large multi-subunit protein complexes. BRCA1 can also play roles not only in DNA repair, but in transcription, ubiquitination, transcriptional regulation, and other cell functions.[30, 31]
B) BRCA2. The BRCA2 gene and protein product, similar to BRCA1, are tumor suppressors referred to as caretaker genes/ proteins found in all humans and primates. The BRCA2 gene is referred to as the “breast cancer type 2 susceptibility gene,” responsible for repairing damaged DNA and chromosomal damage, and inducing cell death in cells where DNA cannot be repaired.[31] The gene is located on the long q-arm of chromosome-13 encoding a protein of 3,418 AAs. Some functions of BRCA2 and BRCA1 are interrelated, even though their molecular structures differ in size. BRCA2 binds to single-strand DNA and directly interacts with the recombinase enzyme RAD51 to stimulate strand invasion, which is a vital step of homologous recombination.[32] PALB2, a partner and localizer of BRCA2, functions synergistically with BRCA2 by linking to a piccolo protein to further promote strand invasion. [33] Like BRCA1, the BRCA2 protein can regulate the activity of other genes and play multiple roles during development.
C) FANCD2, FANC1.The Fanconi anemia (FA) genes comprise a total complementation groups of 19 genes, inherited in an autosomal recessive manner. The FANCD2 gene is located on chromosome 3p with 1,328 AAs, while FANC1 has been localized on chromosome 15q26 having 1,451 AAs. FA genes respond to DNA damage to repair corrupted DNA and protect against chromosome instability.[27] The DNA damage repaired by FA genes encompasses broken and misshapen chromosomes, broken chromatids, and triradial and quadri-radial structures. The lack of DNA repair allows mitosis to proceed with corrupted DNA and enhances damaged cell survival, thus increasing genomic instability. Several components of the FA-DNA repair pathway are the FANCD2-FANC1 heterodimer, the FANCD1-BRCA2 complex, and the BRCA2-interacting protein-1 dimer.[34] In response to DNA damage, the FA protein complexes are activated by the AT kinase and the AT-RAD3-related kinase (ATR).[35] The activated FA protein complexes function as E3 ubiquitin ligases which monoubiquitinate the FAND2/ FANC1 heterodimer. This protein complex then translocates to the chromatin fraction where it combines with other FANC proteins at damaged nuclear replication points.[36] Mutated FA complementation proteins have been linked to the DNA damage/repair at the G2/M checkpoint response during cell cycle progression. However, an absence of G2/M transition arrest can occur with unrepaired double stranded breaks bypassing the checkpoint (CHK1) inhibition at the G2 cell cycle phase.
D) Nibrin.The protein nibrin (NBN) is a 754 AA protein whose gene is located on chromosome 8q21. NBN is a cell cycle regulatory factor, associated with the repair of doublestranded breaks which pose the threat of serious damage to the genome.[37, 38] NBN is associated with the BRCA1/RAD50- containing complex and plays a role in the cellular response to DNA damage and the maintenance of chromosome integrity. [39] The NBN complex is involved not only in double-strand break repair, but in DNA recombination, maintenance of telomere integrity, cell cycle checkpoint control, and meiosis. The complex containing NBN displays single-strand nuclease activity and is involved in control of intra-S phase as well as G1 and G2 checkpoints. The NBN gene is the root cause of the Nijmegen breakage syndrome and related human disorders.
E) The DNA Kinases.The other DNA repair kinases involved in ataxia telangiectasia-mutated ATM, ABL, RAD-related kinases, DNA-PKs, and cell cycle checkpoint kinases, together with their AFP-3D locations, are discussed below and listed in Table 1 (see ref. 24). In addition, the kinase activities of other DNA, cell cycle, and checkpoint protein interactions with AFP-3D are listed in Table 2. Although modest in effect, these kinase assays demonstrate and confirm the interactions of AFP-3D with many DNA and cell cycle-related enzymes.
Table 1. The DNA-repair protein kinases involved in ataxia telangiectasia and RAD-related kinases are displayed according to their properties. The AFP amino acid sequences that interact with these kinases are shown in the right column.
| DNA Kinase | NCBI Accession # | Amino Acid
Length |
Molecular Mass (Kd) | Catalytic Kinase Type | Potential HAFP Amino Acid Binding Sites |
| 1) Ataxia telangiectasia mutated (ATm) | Q13315
AAB65827 NP_000042 |
3065 | 348,395 | P13/P14 kinase, FAT domain, Ser/Thr/tyr, checkpoint kinase | 399GLEEQKY
429NAFLVAYT 449AITRKMAA 461CCQLSEDK 485CIRHEMTP 508RPCFSSLV 529DKFIFHKD 565AFSDDKFI 577GLLEKCCQ |
| 2) Ataxia telangiectasia and RAD3-related (ATR) | CAA70298
Q13535 NP_001175 |
2644 | 300,454 | P13 kinase, Ser/Thr/tyr, RNA helicase, DNA repair protein | 426YYLQNAFL
429NAFLVAYT 444SELMAITR 500CTSSYANR 504YANRRPCF 529DKFIFHKD 549KQEFLINL |
| 3) DNA-dependent protein kinase DNA-PK (ATm-related)
DNA-PKCS |
P78527
|
4128 | 469,090 | DNA-PK, P13K, Ser/Thr/tyr, molecular sensor of DNA damage | 421KLFEYYLQ
429NAFLVAYT 461CCQLSEDK 481IGHLCIRH 508RPCFSSLV |
| 4) Serine/threonine protein kinase (CHK2) checkpoint | 096017 | 543 | 60,453 | Required for checkpoint-mediated cell cycle arrest, activation of DNA repair, apoptosis, and negative regulation or cell cycle | 436KKAPQLTS
440QLTSSELM |
| 5) c-ABL1 Abelson murine leukemia oncogene homolog-1, partner with Philadelphia chromosome | P00519 | 1130 | 125,804 | Cytoplasmic and nuclear protein tyrosine kinase, DNA binding, cell cycle function | 421KLGEYYA
477ADIIIGHL 500CTSSYANR 597QKLISKTR |
Ser = serine; Thr = threonine; tyr = tyrosine;
PK = protein kinase; RAD3 = DNA helicase domain; FAT = Focal Adhesion Tyrosine Kinase
*Ataxia telangiectasia mutated (ATm) is P13/P14 kinase FAT domain Ser/Thr/tyr, checkpoint kinase AFP = alpha-fetoprotein.
Table 2. The percent of kinase enzyme activity‡ following AFP-3D GIP peptide treatment is listed below. The control assay was 100% and the inhibition or enhancement is listed as percent activity of the control assays performed in IC50 titration curves. Note that AFP-3D kinase inhibition is associated with Ser/Thr kinases while Tyr kinases are associated mostly with enhancements.
| I. Kinase Enzyme Name | Type c-SRC 2,3* | Inhibition Percent ± SD | Activity |
| 1) ASK-1 | Ser/Thr | 28 ± 4 | Oxidative stress, MAP-kinases |
| 2) cdK3/cyclin E | Ser/Thr | 18 ± 0 | G1 → S cell cycle control |
| 3) cdK5/p35 | Ser/Thr | 28 ± 3 | G2 → M transition, histone binding |
| 4) MKK7B | Ser/Thr | 18 ± 9 | G2-M arrest, MAP kinase |
| 5) MSK2 | Ser/Thr | 18 ± 1 | Stress, chromatin binding |
| 6) MST1 | Ser/Thr | 17 ± 14 | Histone, telomerase-related |
| 7) PKCa | Ser/Thr | 23 ± 2 | Cell cycle checkpoint |
| II. Kinase Enzyme Name | Type SRC 2,3 | Enhancement Percent ± SD | Activity |
| 1) EpHA4 | Tyr | 30 ± 10 | Neurons, cell migration |
| 2) EpHB4 | Tyr | 19 ± 2 | Cell migration, vascular development |
| 3) Erb-B4 | Tyr | 18 ± 2 | Epidermal growth factor signal, mitogenesis |
| 4) EGFR1 | Tyr | 22 ± 4 | Epidermal growth factor receptor, DNA synthesis |
| 5) FGFR2 | Tyr | 18 ± 1 | Adhesion-related mitogenesis, diff. |
| 6) IGF-1R | Ser/Thr | 18 ± 1 | Insulin growth factor, cell division |
| 7) Met | Tyr | 21 ± 0 | Proto-oncogene tumor growth |
* Ser/Thr – Serine/thyronine kinase; Tyr – tyrosine kinase;
c-Src – a non-receptor kinase protein of the Ser/Thr or tyrosine type that phosphorylates these residues in other proteins
‡ The kinase activity screen for AFP-3D peptides was performed via the commercial “kinase profiler” by the Upstate Biosignaling Corp., Dundee Technology Park, Dundee, United Kingdom.
Computer Analysis of AFP-3D Interaction with DNA-Repair Proteins
The third domain of AFP is known to interact with a myriad of proteins and compounds including hydrophobic ligands, receptors, and cytoplasmic binding proteins. Previous publications from the author (GJM) and others have confirmed and verified these reports (see above). These interacting agents include fatty acids, steroids (estrogens), retinoids, cation channels, cell cycle proteins, and chemokine, mucin, and scavenger receptors.[8, 9, 11, 13, 23] These interacting agents have previously been mapped to the aminoterminal, middle, and carboxy-terminal portions of the AFP-3D. [8, 13] The amino terminal portion of AFP-3D is known to interact with fatty acids, estrogens, steroids, retinoids and lysophospholipids, while the middle and carboxy-terminus portions react with scavenger, mucin, and cation channels. Lastly, the carboxy-terminal fragment displays interaction sites with cell cycle proteins, cation channels, chemokine receptors, and dimerizing proteins. Data from the present study now reveal that DNA repair proteins represent additional binding/interaction sites on the AFP-3D fragment.
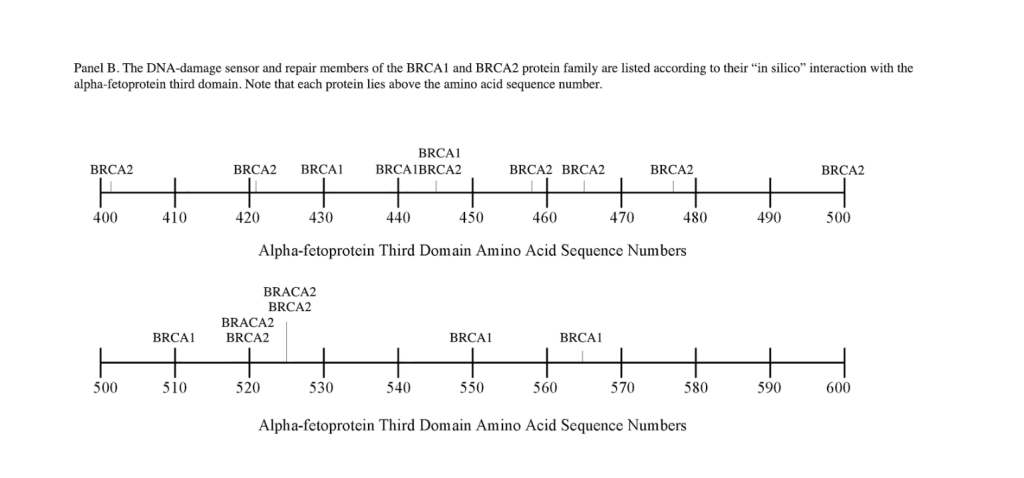
The DNA repair protein interaction sites similar to previous ligands and receptors, were distributed in patterns of interspaced clustered groups throughout the AFP third domain. The BRCA1/BRCA2 sites were heavily distributed on the first half of the AFP-3D fragment from AA #420 to 500, with another cluster localized at AA #510 to 530 with outliers at AA #550 to 565 (Figure 1). Figure 1, Panel A shows that BRCA1/BRCA2 sites were localized within the hydrophobic ligand binding and lysophospholipid receptor subdomain. This site further overlaps with the Growth Inhibitory Peptide (GIP) and cell cycle protein segment, together with the anterior portion of the scavenger receptor sites. The BRCA1/BRCA2 interacting sites at AA #510 to 530 were found to be localized among the cell cycle and cation channel proteins and the mucin/chemokine receptor binding sites.
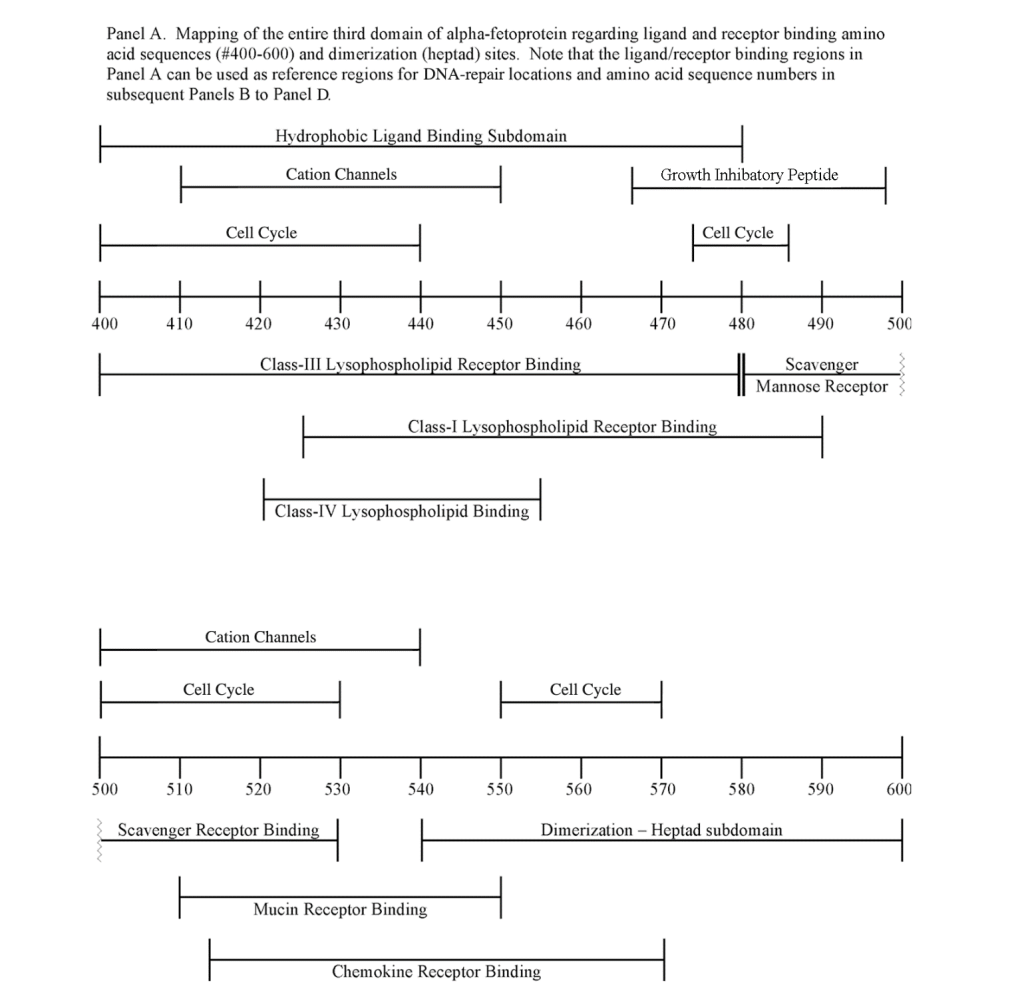
The FANC1/FANCD2 interaction sites were scantily localized at AA #430 to 460 and AA #480 to 490 in contrast, the FANC proteins were heavily distributed within the second half of the AFP-CD segment extending from AA #500 to 580 (Figure 1). As shown with the BRCA1/BRCA2 proteins, the FANC proteins localized among the hydrophobic ligand-binding areas and the GIP segment in the first half of the AFP-3D segment. However, in the second half of AFP-3D, the FANC protein sites were distributed among the scavenger, mucin, and chemokine receptors in addition to the cation channel protein binding/interaction sites.
The third DNA repair protein, nibrin, was localized to the AFP- 3D, largely in the second half of the AFP third domain from AA #500 to 530 and AA #565 to 580, with an outlier at AA #480. These regions correspond largely to the scavenger, mucin, and chemokine receptor regions of AFP-3D together with the corresponding protein interaction sites at the GIP AA segment.
Proposed Relationship of DNA Repair Proteins with the AFP-3D Hydrophobic Binding and Receptor Sites
As described above, the DNA repair proteins localization sites were found to coincide with previously identified hydrophobic ligand and receptor binding sites. Prior reports in the literature have described associations and interrelationships that exist between the hydrophobic ligands and the receptors hence, the DNA repair protein pairing localizations may be more than a mere coincidence. For example, the BRCA gene expression is known to be significantly reduced in human (MCF-7) rat mammary tumorigenesis by the supplementation of omega-3 fatty acids (docosahexaenoic acid) in the diet.[40, 41] Prior research showed that BRCA1 acts as a scaffold protein in multiple cellular functions such as transcription, DNA repair, and ubiquitination by interaction with acetyl-CoA carboxylase.[42, 43] BRCA1 is also implicated in novel signaling pathways associated with fatty acid-dependent breast cancer proliferation when associated with supplemented diet fatty acids and ERK1/2, p53-p21 WAF1/CIP1, MAPK, p27 KIP1, and NF-KappaB proteins.[44] The N-3 and N-6 polyunsaturated fatty acids are reported to have differential effects on gene expression of BRCA1 and BRCA2 in human breast cancer cell lines (MCF-7, MDA-MB-231).[45, 46] In contrast, BRCA1 and BRCA2 had no relationships with scavenger receptors and chemokine receptors, as shown in Figure 1 (panels A & B). Moreover, present findings support the association reported in the literature of BRCA1/ BRCA2 DNA-repair proteins with the cell cycle proteins (Figure 1, panels A & B). It was found that the cell cycle proteins were coincident with DNA repair proteins in the localization sites of AA #480 to 490 and AA #510 to 580. Prior studies support this relationship, showing a cyclin-D induced gene amplification and hypermethylation together with CdK12 inhibition in human breast cancer BRCA positive patients.[47-50] In light of the dual localization of mucin receptors and BRCA1/BRCA2 (AA #510 to 530), DNA repair proteins have been studied to determine whether pre- and postoperative CA125 levels are associated with BRCA mutation carriers in ovarian cancer screenings.[51, 52]
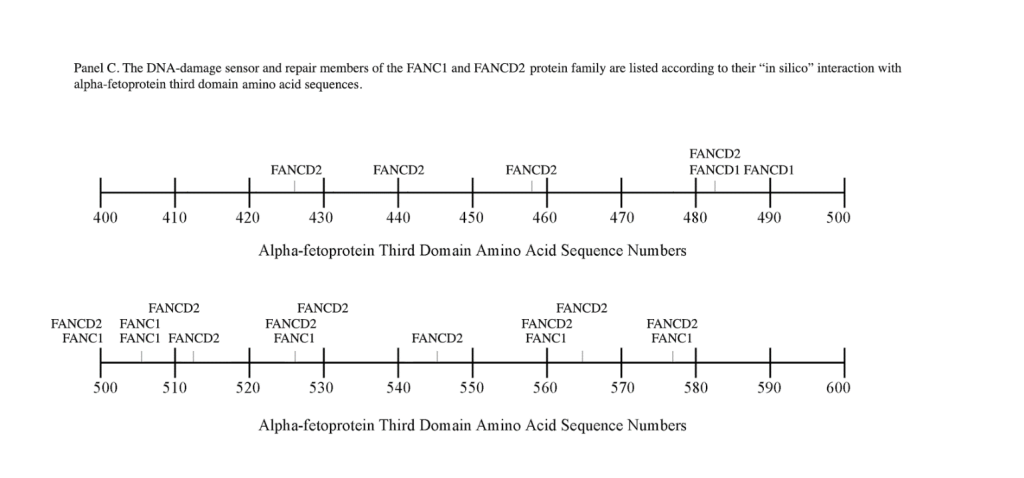

Regarding FANC protein localization with hydrophobic ligand and receptor interaction sites, no associations were found relating DNA repair to either fatty acids, mucin receptors, lysophospholipids or cation channels however, DNA repair interaction sites were observed at AA #481 to 504, a known GIP and chemokine receptor area (Figure 1, panel C). Indeed, one study demonstrated a link between chemokine CXCR5 receptors and FANCA-modulated neddylation pathways involved in membrane targeting and cell mobility.[53] Regarding the nibrin protein interaction sites on AFP-CD, NBN sites were largely localized to the second half of the 3D fragment. Nibrin was found to be localized with the ataxia telangiectasia-mutated (ATM) protein, checkpoint kinase-2, and the RAD-related protein (see Table 1, panels A & D), as well as the BRCA1/BRCA2 proteins all of which contribute to breast cancer susceptibility.[54-56] These protein complexes are involved in the dysfunction of specific DNA double-strand break-repair signaling pathways. Other reports of putative ATM in vitro interaction targets include nibrin, RAD17, PTS, and ATM itself.[57]
Relationship of AFP to DNA Damage and Repair Disorders
The correlation of AFP to DNA damage/repair and chromosome instability disorders is well documented, in part because AFP is a biomarker for both immunodeficiency diseases and anemia disorders.[23, 27] Elevated AFP serum levels have been reported in immunodeficiency disorders such as ataxia telangiectasia (AT) and ataxia ocular apraxia (AOA2). The AOA2 disorder displays aberrant DNA repair proteins, ATM mutated in AT and senataxin in AOA, and ATR in AT and RAD3-related disorders.[58, 59] AFP intracellular levels have also been correlated with intracytoplasmic levels of GADDI53 (growth arrest and DNA damage-inducible gene I53) in vascular smooth muscle cell death.[60]
AT is a chromosomal instability disorder caused by an autosomal recessive gene. AT is characterized by increased cell radio-sensitivity and multiple chromosomal aberrations in the DNA of immune cells these include gaps, breaks, dicentrics, and multiple-radial configurations. Most patients (90%) with AT display high serum AFP levels, which can range from 30 to 400 ng/mL.[61-63] ATM interaction sites on AFP-3D were presently localized at AA #429 to 485, AA #500 to 506, and AA #560 to 580 (Figure 1). Patients with AT also exhibit aberrant cell checkpoint proteins that allow continuation through the cell cycle, despite DNA breaks that require repair before the next replication stage occurs. As a consequence, AT patients show a propensity to develop cancer later in life.
Once cloned, the ATM protein was found to be a kinase that shares sequence homology with RAD-3, a kinase that regulates passage (via checkpoints) through the cell cycle after DNA damage has occurred. ATM is also involved with the PI3-kinase signal transduction pathway. [64] The RAD-3 kinase has been cloned and named the AT-RAD3- related (ATR) kinase. The ATR kinase was presently localized on AFP- 3D in two clusters, one at AA #426 to 444 and the other at AA #500 to 539. The former cluster lies directly within the hydrophobic ligand binding region, the cation channel, and the lysophospholipid receptor interaction sites. The latter site was localized among the scavenger, mucin, and chemokine receptor and cell cycle interaction sites. It is of interest that the latter site coincides with cell cycle-associated checkpoint proteins during cell cycle progression.[65, 66] Non-mutated AT/ATR protein kinases sense the presence of double stranded DNA damage and are known to mediate an appropriate repair response. Lastly, a phosphoinositol kinase-3 (PI3-kinase) that associates with the ATM/ATR protein complex, termed DNA-PKCS (Table 1), is a required kinase associated with DNA repair of non-homologous end joining, whose absence results in chromosomal aberrations.[67] The DNA-PKCS interaction sites were localized on AFP-3D at AA #420 to 481, coinciding with hydrophobic binding and the cation channel sites, as well as cell-cycle associated and lysophospholipid receptor interaction sites (Table 1, Figure 1, panel A).
Fanconi’s Anemia (FA) is another DNA-damage/repair disorder associated with both chromosome instability and elevated serum AFP levels both in early infancy and adults. FA represents a progressive, autosomal recessive disorder that exhibits DNA damage, chromosomal breaks, bone marrow failure, and a predisposition to malignancies. [68] Cells from FA patients further display a delay and/or arrest in the G2-to-mitotic transition phase of the cell cycle. As discussed earlier, the FANC proteins represent a complementation group made up of multiple different proteins. However, the present study only addresses FANC1 and FAND2. The origin and source of elevated AFP in FA is presently unknown, since liver dysfunction abnormalities and disease (cancer) are not involved with FA. The author.[27] has suggested that the origin of AFP synthesis and production may lie in the existence of three stem-progenitor cell types present in adult bone marrow namely, fetal hepatic stem/progenitor cells and intrinsic hematopoietic stem/ progenitor cells (HSPC). A third stem bone marrow cell termed the “mesenchymal stem cell” is capable of migrating to the liver and differentiating into hepatocyte-like stem cells following hepatic failure, regeneration, and liver transplantation. Interestingly, the classical hepatic oval cell population surrounding bile ducts are the actual cells that secrete AFP and express the immature stem cell markers CD34 and CD45. Thus, small to moderate amounts of AFP production/secretion could occur in acutely anemic bone marrow with no detectable liver damage, dysfunction, or disease in the FA patient.
Concluding Remarks
It is well-established in the literature that the AFP-3D houses subdomain interaction (interface) sites for a myriad of ligands, receptors, cation channels, cell cycle proteins most recently, DNA damage/repair proteins have been added to this list.[8, 9, 13] These third domain protein interaction sites were first detected by computer analysis, and then verified in cell-based assays, microarray analysis, in vitro cell cultures, and in vivo animal (xenograft) models. For example, an RNA global microarray analysis using AFP-3D derived peptides (see GIP sequence, Figure 1) demonstrated that DNA repair proteins do indeed react with the AFP-GIP amino acid sequences #464-496.[14] The microarray analysis showed that the GIP AA sequences downregulated the mRNA of FANCD2 and up-regulated BRCA1 and RAD54c (Table 2). In addition, histone-1-H4g (DNA-repair) and checkpoint suppressor-1 were greatly downregulated, while multiple DNA repair proteins were modestly upregulated in proteins such as BRCA1 ring domain and RAD5/c (Table 3). Hence, published data confirms that AFP AA sequences on AFP-3D can interact and regulate the RNA of DNA repair proteins in conjunction with cell cycle progression proteins. It is conceivable that the different ligands, proteins, channels, and receptors could react simultaneously in combination with, or in competition with, direct or adjacent interaction sites.
Table 3. Global RNA microarray data following AFP-derived peptide treatment: Transcripts displaying 1.0 or larger log fold (log base 2.0) decrease for genes associated with cell division and proliferation processes, ubiquitization, and DNA repair proteins obtained from human MCF-7 breast cancer cells in vitro.*
| GENE PROTEIN TITLE | ||
| Part I. Cell Cycle Regulation/DNA Repair | FOLD DECREASE (–) |
CELL FUNCTION |
| 1. Checkpoint suppressor-1 (CHES1)(FOXN3) | –9.2 | S-phase checkpoint |
| 2. Cyclin-E** | –4.6 | Regulates G-S transition |
| 3. Transcription Dp-1 (TFDP1) | –4.3 | G1 to S-phase transition |
| 4. CDC20 cell division homolog | –4.3 | Regulation of cell cycle |
| 5. Histone-1, H4g (HIST1H4G) | –3.2 | DNA repair/replication |
| 6. Fanconi anemia-D2 (FRANCD2) | –2.0 | DNA repair/synthesis |
| 7. TAF-1-like polymerase | –0.8 | DNA repair/synthesis |
| 8. Excision repair cross complement | –0.5 | DNA repair |
| Part II. Cell Cycle Phase Transition and DNA Repair | FOLD INCREASE (+) |
CELL FUNCTION |
| 1. RAD5/c | +1.5 | DNA repair |
| 2. Polymerase DNA directed kappa | +0.5 | DNA repair |
| 3. BRCA1 associated ring domain | +0.4 | DNA repair |
| 4. Methyl GpG binding domain | +0.4 | DNA repair |
| 5. CDC2 cell division C2 | +0.4 | G1-S, G2-M transition |
| 6. RAD54 homolog-B | +0.4 | DNA repair |
| 7. Ubiquitin-specific protease-1 | +0.3 | DNA repair |
| 8. S-phase kinase-associated protein-2 | +0.3 | G1-S-phase transition |
* Expression of 716 transcripts was significantly altered in MCF-7 cells after 8 days of treatment with GIP as compared to treatment with the scrambled peptide. Four-hundred thirty RNAs were down-regulated, while 286 RNAs were upregulated.
** Real time PCR. Collaborative data was provided by Kathleen Arcaro, University of Massachusetts, Amherst, MA (14).
The interaction sites described in this report obviously have links to other proximal and/or distal sites along the AFP third domain fragment. As discussed above, literature-based reports have documented that DNA-repair proteins do in fact interact with cell cycle checkpoint proteins to arrest cell cycle progression.[69] Furthermore, BRCA1/BRCA2 can act as scaffold proteins in the ubiquitinization of cell cycle proteins through a proteasomal pathway [70] As shown above, BRCA1/BRCA2 were found to be associated with fatty acid-dependent breast cancer growth.[40-43] Prior reports have further demonstrated that computer-derived cation channel interaction sites were localized at hydrophobic ligand as well as lysophospholipid receptor binding sites.[71] Thus, it is plausible that the clustered localization of channel and cell cycle proteins with DNA repair proteins could be physiologically relevant.
In the present report, evidence was presented that several mutated proteins of the DNA-damage/repair pathways are associated with cancer susceptibility, tumorigenesis, and enhancement of tumor progression, most notably in breast and ovarian cancer, but in other cancers as well. The BRCA and FA-related protein mutations leading to anemia subjects these patients to develop tumors later in life.[72] A significant connection of AFP to FA-mutated DNA repair proteins lies in the elevations of serum AFP in such anemic patients. There are further correlations with breast cancer and its associated BRCA1/ BRCA2 mutated proteins. Sarcione et al. has reported that a circulating bound form of serum AFP, as opposed to free circulating AFP, exists in some female breast cancer patients. This bound form of AFP could be experimentally released by high KCl solutions and measured by immunological assays.[73] Although the bound entity is not known, an IgM molecule has been similarly reported to complex with serum AFP as a bound form.[73] It has also been reported that an intracytoplasmic non-secreted form of AFP is present in normal cells, as well as cancer cells. The non-secreted cytoplasmic AFP (cAFP) form has been shown to participate in kinase regulation, transcription, apoptosis, nuclear hormone binding, transnuclear passage, and regulation of nuclear gene expression.[74, 75] One mechanism of this AFP interaction in cytoplasmic protein activities involves the heterodimerization of AFP with proteins such as cytoplasmic caspases and retinoic acid nuclear receptors.[76] (Figure 1, panel A). Thus, these observations support the contention that cAFP reacts with intracytoplasmic proteins such as the BRCA1/BRCA2, FANC1/FANCD2, nibrin, ATM, and ATR, as suggested by the present report. Furthermore, the reported observations of interaction of AFP-3D with cell cycle proteins, together with the DNA-repair proteins association with cell cycle checkpoints, allows for speculation that AFP could mask, interfere, enhance, or interpose itself into the DNA-repair process of the cell cycle checkpoint regulation pathway. The RNA microarray analyses (Table 3) are consistent with this supposition. The above studies beg the question of whether cAFP (by means of the third domain) is a prime regulatory factor in the overall scheme of DNA repair during cell cycle progression.
Acknowledgements: The author thanks Kathleen Arcaro, University of Massachusetts, Amherst, MA for providing data on the RNA microarray analysis. The author also wishes to thank Mr. Andrew Bentley (Wadsworth Center Photography and Medical Illustration Department) for his expertise in producing the figures and graphic art illustrations and Ms. Tracy Godfrey for her typing, corrections, revisions, and processing of this manuscript.
Competing interests: The author declares that he has no competing interests.
Funding information: The author has no such involvement
Abbreviations
AA – amino acid
AFP – alpha-fetoprotein 3D – third domain
DNA – deoxyribonucleic acid
DDSR – DNA damage-sensing and repair
PI3K – phosphoinositol kinase
mTOR – mechanistic target of rapamycin
GAAD153 – growth arrest and DNA damage-inducible-protein-153
PTEN – phosphatase and tensin homolog
ATM – ataxia telangiectasia-mutated
FA – Fanconi’s anemia
CHK – checkpoint
BRCA – breast cancer
RAD (ATR) – AT-related repair of DNA
NBN – nibrin
ABL – Abelson leukemia oncogene
GIP – growth inhibitory peptide
CdK-cyclin dependent kinase
PTS – 6-pyruvoyltetrahydropterin synthase
AOA – ataxia ocular apraxia
PKC – protein kinase- C.
References
- Mizejewski GJ (2002) Biological role of alpha-fetoprotein in cancer: prospects for anticancer therapy. Expert Rev Anticancer Ther.
- Mizejewski GJ (2004) Biological roles of alpha-fetoprotein during pregnancy and perinatal development. Exp Biol Med (Maywood) 229: 439-463. [crossref]
- Mizejewski GJ (2001) Alpha-fetoprotein structure and function: relevance to isoforms, epitopes, and conformational variants. Exp Biol Med (Maywood). 2001 May.
- Mizejewski GJ (1997) alpha-fetoprotein as a biologic response modifier: relevance to domain and subdomain structure. Proc Soc Exp Biol Med 215: 333-362. [crossref]
- Naidu S, Peterson ML, Spear BT (2010) Alpha-fetoprotein related gene (ARG): a new member of the albumin gene family that is no longer functional in primates. Gene 449: 95-102. [crossref]
- Herve F, Rajkowski KM, Martin MT, Dessen P, et al. (1986) Drug-binding properties of rat alpha-foetoprotein. Specificities of the phenylbutazone-binding and warfarin-binding sites. Biochem J.
- Li MS, Li PF, He SP, Du GG, Li G (2002) The promoting molecular mechanism of alpha-fetoprotein on the growth of human hepatoma Bel7402 cell line. World J Gastroenterol 8: 469-475. [crossref]
- Mizejewski GJ (2015) The alpha-fetoprotein third domain receptor binding fragment: in search of scavenger and associated receptor targets. J Drug Target.
- Mizejewski GJ (2013) Review of the adenocarcinoma cell surface receptor for human alpha-fetoprotein; proposed identification of a widespread mucin as the tumor cell receptor. Tumour Biol.
- Atemezem A, Mbemba E, Marfaing R, Vaysse J, Pontet M, et al. (2002) Human alpha-fetoprotein binds to primary macrophages. Biochem Biophys Res Commun 296: 507-514. [crossref]
- Mizejewski GJ (2015) Nonsecreted cytoplasmic alpha-fetoprotein: a newly discovered role in intracellular signaling and regulation. An update and commentary. Tumour Biol.
- Li M, Li H, Li C, Zhou S, et al. (2009) Alpha fetoprotein is a novel protein-binding partner for caspase-3 and blocks the apoptotic signaling pathway in human hepatoma cells. Int J Cancer.
- Mizejewski GJ (2016) The alpha-fetoprotein (AFP) third domain: a search for AFP interaction sites of cell cycle proteins. Tumour Biol.
- Mizejewski GJ (2011) Mechanism of Cancer Growth Suppression of Alpha-Fetoprotein Derived Growth Inhibitory Peptides (GIP): Comparison of GIP-34 versus GIP-8 (AFPep). Updates and Prospects. Cancers (Basel).
- Posypanova GA, Gorokhovets NV, Makarov VA, Savvateeva LV, et al. (2008) Recombinant alpha-fetoprotein C-terminal fragment: the new recombinant vector for targeted delivery. J Drug Target.
- Posypanova GA, Makarov VA, Savvateeva MV, Bereznikova AV, et al. (2013) The receptor binding fragment of alpha-fetoprotein is a promising new vector for the selective delivery of antineoplastic agents. J Drug Target.
- Yabbarov NG, Posypanova GA, Vorontsov EA, Obydenny SI, et al. (2013) A new system for targeted delivery of doxorubicin into tumor cells. J Control Release.
- Sharapova OA, Pozdnykova NV, Laurinavichyute DK, Yurkova MS, et al. (2010) High-efficient expression, refolding and purification of functional recombinant C-terminal fragment of human alpha-fetoprotein. Protein Expr Purif.
- Sharapova OA, Iurkova MS, Andronova SM, Fedorov AN, et al. (2011) [High-efficient renaturation of immobilized recombinant C-terminal fragment of human alpha-fetoprotein]. Prikl Biokhim Mikrobiol.
- Sharapova OA, Pozdniakova NV, Laurinavichiute DK, Iurkova MS, et al. (2010) [Purification and characterization of recombinant human alpha-fetoprotein fragment, corresponding to the C-terminal structural domain]. Bioorg Khim.
- Carter DC, He XM, Munson SH, Twigg PD, Gernert KM, et al. (1989) Three-dimensional structure of human serum albumin. Science 244: 1195-1198. [crossref]
- Osmond RI, Das S, Crouch MF (2010) Development of cell-based assays for cytokine receptor signaling, using an AlphaScreen SureFire assay format. Anal Biochem 403: 94-101. [crossref]
- Mizejewski G (2014) Alpha-fetoprotein as a biomarker in immunodeficiency diseases: relevance to ataxia telangiectasia and related disorders. J Immunodeficiency and Disorders.
- Duncan JA, Reeves JR, Cooke TG (1998) BRCA1 and BRCA2 proteins: roles in health and disease. Mol Pathol 51: 237-247. [crossref]
- Friedenson B (2007) The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers. BMC Cancer 7: 152. [crossref]
- Yoshida K and Miki Y (2004) Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci.
- Lakhi NA and Mizejewski GJ (2016) Alpha-fetoprotein and Fanconi Anemia: Relevance to DNA Repair and Breast Cancer Susceptibility. Fetal Pediatr Pathol.
- Starita LM, Parvin JD (2003) The multiple nuclear functions of BRCA1: transcription, ubiquitination and DNA repair. Curr Opin Cell Biol 15: 345-350. [crossref]
- Wang Y, Cortez D, Yazdi P, Neff N, et al. (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev.
- Paterson JW (1998) BRCA1: a review of structure and putative functions. Dis Markers 13: 261-274. [crossref]
- Henderson BR (2005) Regulation of BRCA, BRCA2 and BARD1 intracellular trafficking. Bioessays 27: 884-893. [crossref]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, et al. (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22: 719-729. [crossref]
- Buisson R, Dion-Cote AM, Coulombe Y, Launay H, et al. (2010) Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol.
- Kondo N, Takahashi A, Mori E, Noda T, et al. (2011) FANCD1/BRCA2 plays predominant role in the repair of DNA damage induced by ACNU or TMZ. PLoS One.
- O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, et al. (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet.
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, et al. (2007) Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair.
- Desai-Mehta A, Cerosaletti KM and Concannon P (2001) Distinct functional domains of nibrin mediate Mre11 binding, focus formation, and nuclear localization. Mol Cell Biol.
- Chen YC, Su YN, Chou PC, Chiang WC, Chang MC, et al. (2005) Overexpression of NBS1 contributes to transformation through the activation of phosphatidylinositol 3-kinase/Akt. J Biol Chem 280: 32505-32511. [crossref]
- Zhong Q, Chen CF, Li S, Chen Y, Wang CC, et al. (1999) Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 285: 747-750. [crossref]
- Jourdan ML, Mahéo K, Barascu A, Goupille C, De Latour MP, et al. (2007) Increased BRCA1 protein in mammary tumours of rats fed marine omega-3 fatty acids. Oncol Rep 17: 713-719. [crossref]
- Bernard-Gallon DJ, Vissac-Sabatier C, Antoine-Vincent D, Rio PG, et al. (2002) Differential effects of n-3 and n-6 polyunsaturated fatty acids on BRCA1 and BRCA2 gene expression in breast cell lines. Br J Nutr.
- Moreau K, Dizin E, Ray H, Luquain C, Lefai E, et al. (2006) BRCA1 affects lipid synthesis through its interaction with acetyl-CoA carboxylase. J Biol Chem 281: 3172-3181. [crossref]
- Hilakivi-Clarke L, Olivo SE, Shajahan A, Khan G, Zhu Y, et al. (2005) Mechanisms mediating the effects of prepubertal (n-3) polyunsaturated fatty acid diet on breast cancer risk in rats. J Nutr 135: 2946S-2952S. [crossref]
- Menendez JA, Mehmi I, Atlas E, Colomer R, et al. (2004) Novel signaling molecules implicated in tumor-associated fatty acid synthase-dependent breast cancer cell proliferation and survival: Role of exogenous dietary fatty acids, p53-p21WAF1/CIP, ERK1/2 MAPK, p27KIP, BRCA, and NF-kappaB. Int J Oncol.
- Kachhap SK, Dange PP, Santani RH, Sawant SS, et al. (2001) Effect of omega-3 fatty acid (docosahexanoic acid) on BRCA1 gene expression and growth in MCF-7 cell line. Cancer Biother Radiopharm.
- Bernard-Gallon DJ, Maurizis JC, Rio PG, Bougnoux P, Bignon YJ (1998) Effects of monounsaturated and polyunsaturated fatty acids (omega-3 and omega-6) on Brca1 protein expression in breast cell lines. J Natl Cancer Inst 90: 1234-1235. [crossref]
- Locke I, Kote-Jarai Z, Fackler MJ, Bancroft E, et al. (2007) Gene promoter hypermethylation in ductal lavage fluid from healthy BRCA gene mutation carriers and mutation-negative controls. Breast Cancer Res.
- Johnson SF, Cruz C, Greifenberg AK, Dust S, et al. (2016) CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep.
- Osin P, Gusterson BA, Philp E, Waller J, et al. (1998) Predicted anti-oestrogen resistance in BRCA-associated familial breast cancers. Eur J Cancer.
- Plevova P, Cerna D, Balcar A, Foretova L, Zapletalova J, et al. (2010) CCND1 and ZNF217 gene amplification is equally frequent in BRCA1 and BRCA2 associated and non-BRCA breast cancer. Neoplasma 57: 325-332. [crossref]
- Fields MM, Chevlen E (2006) Ovarian cancer screening: a look at the evidence. Clin J Oncol Nurs 10: 77-81. [crossref]
- Tierney BJ, McCann GA, Cohn DE, Eisenhauer E, et al. (2012) HO-3867, a STAT3 inhibitor induces apoptosis by inactivation of STAT3 activity in BRCA1-mutated ovarian cancer cells. Cancer Biol Ther.
- Renaudin X, Guervilly JH, Aoufouchi S and Rosselli F (2014) Proteomic analysis reveals a FANCA-modulated neddylation pathway involved in CXCR5 membrane targeting and cell mobility. J Cell Sci.
- Uzunoglu H, Korak T, Ergul E, Uren N, et al. (2016) Association of the nibrin gene (NBN) variants with breast cancer. Biomed Rep.
- Cerosaletti KM and Concannon P (2003) Nibrin forkhead-associated domain and breast cancer C-terminal domain are both required for nuclear focus formation and phosphorylation. J Biol Chem.
- Damiola F, Pertesi M, Oliver J, Le Calvez-Kelm F, et al. (2014) Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res.
- Keimling M, Volcic M, Csernok A, Wieland B, et al. (2011) Functional characterization connects individual patient mutations in ataxia telangiectasia mutated (ATM) with dysfunction of specific DNA double-strand break-repair signaling pathways. FASEB J.
- Suraweera A, Becherel OJ, Chen P, Rundle N, et al. (2007) Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol.
- Asaka T, Yokoji H, Ito J, Yamaguchi K, et al. (2006) Autosomal recessive ataxia with peripheral neuropathy and elevated AFP: novel mutations in SETX.
- Igase M, Okura T, Nakamura M, Takata Y, et al. (2001) Role of GADD153 (growth arrest- and DNA damage-inducible gene 153) in vascular smooth muscle cell apoptosis. Clin Sci (Lond).
- Waldmann TA, McIntire KR (1972) Serum-alpha-fetoprotein levels in patients with ataxia-telangiectasia. Lancet 2: 1112-1115. [crossref]
- Ishiguro T, Taketa K, Gatti RA (1986) Tissue of origin of elevated alpha-fetoprotein in ataxia-telangiectasia. Dis Markers 4: 293-297. [crossref]
- Stray-Pedersen A, Borresen-Dale AL, Paus E, Lindman CR, Burgers T, et al. (2007) Alpha fetoprotein is increasing with age in ataxia-telangiectasia. Eur J Paediatr Neurol 11: 375-380. [crossref]
- Lavin MF, Khanna KK, Beamish H, Spring K, et al. (1995) Relationship of the ataxia-telangiectasia protein ATM to phosphoinositide 3-kinase. Trends Biochem Sci.
- Dierov J, Dierova R and Carroll M. BCR/ABL translocates to the nucleus and disrupts an ATR-dependent intra-S phase checkpoint. Cancer Cell. 2004 Mar.
- Dart DA, Adams KE, Akerman I and Lakin ND (2004) Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J Biol Chem.
- Hartley KO, Gell D, Smith GC, Zhang H, et al. (1995) DNA-dependent protein kinase catalytic subunit: a relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product.
- Yang YG, Herceg Z, Nakanishi K, Demuth I, et al. (2005) The Fanconi anemia group A protein modulates homologous repair of DNA double-strand breaks in mammalian cells.
- Nilsson I, Hoffmann I (2000) Cell cycle regulation by the Cdc25 phosphatase family. Prog Cell Cycle Res 4: 107-114. [crossref]
- Carlucci A, D’Angiolella V2 (2015) It is not all about BRCA: Cullin-Ring ubiquitin Ligases in ovarian cancer. Br J Cancer 112: 9-13. [crossref]
- Mizejewski GJ (2016) Review of the Third Domain Receptor Binding Fragment of Alpha-fetoprotein (AFP): Plausible Binding of AFP to Lysophospholipid Receptor Targets. Current Drug Targets.
- L, Liu P, Evans TC, Jr. and Ettwiller LM (2017) DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification.
- Sarcione EJ and Biddle W (1987) Elevated serum alpha fetoprotein levels in postmenopausal women with primary breast carcinoma. Dis Markers.
- Beneduce L, Castaldi F, Marino M, Tono N, et al. (2004) Improvement of liver cancer detection with simultaneous assessment of circulating levels of free alpha-fetoprotein (AFP) and AFP-IgM complexes. Int J Biol Markers.
- Jingting J, Changping W, Ning X, Yibei Z, et al. (2009) Clinical evaluation of serum alpha-fetoprotein-IgM immune complexes on the diagnosis of primary hepatocellular carcinoma. J Clin Lab Anal.
- Li M, Li H, Li C, Guo L, et al. (2009) Cytoplasmic alpha-fetoprotein functions as a co-repressor in RA-RAR signaling to promote the growth of human hepatoma Bel 7402 cells. Cancer Lett.