Abstract
Chagas disease, a parasitic, endemic infection affects about 10–15 million of people all over Latin America. In addition, Chagas disease has an increased global impact in nonendemic areas due to immigration patterns. More than 10, 000 deaths are caused by this disease each year, and nearly 70 million people are susceptible to infection. Although Chagas disease was identified more than 100 years ago, current therapeutic options for this condition are limited mainly to two ineffective drugs, benznidazole and nifurtimox. These two drugs are not optimal as currently applied because they show substantial toxicity, require long courses of administration and have inconsistent efficacy. In light of these problems, safer and more effective therapeutic options for patients with Chagas disease are clearly needed. Many candidate drugs are currently being tested. Among them, cruzapain inhibitors are considered promising agents that have been shown to have potential for more effective treatment of the chronic form of Chagas disease. In this work we review briefly the most significant advance in the design of new molecules able to kill Trypanosoma cruzi via an inhibition of the enzyme cruzain (also referred to as cruzipain, the full-length native enzyme).
Keywords
Chagas disease; Anti-chagasic drugs; Cruzain; Irreversible cruzain inhibitors; Reversible cruzain inhibitors; Vinyl sulfones; Vinyl nitroalkenes; Amidonitriles; Lipinski’ parameters and in silico study
Introduction
Among 17 Neglected Tropical Diseases (NTDs), Chagas Disease (CD, American trypanosomiasis) has been considered one of the most important tropical protozoan afflictions [1–3]. To treat parasitic infections like CD, there are four general tools, e.g. diagnostics, vector control agents, vaccines and drugs. Nevertheless, these disease-specific technologies are still deficient and have narrow use because of their inadequate efficacies and toxicities, or because they do not avoid reinfection. This also applies to Chagas disease, an important health problem in Central and South America [4, 5]. Since 1908 it is well-known that the CD is caused by infection with the haemoflagellate protozoan Trypanosoma cruzi, which is transmitted in rural areas to humans and other mammals by reduviid bugs such as Rhodnius prolixus, Triatoma dimidiate and Triatoma infestans, so-called kissing bugs, the most important vectors. The latter insects were usually reported in sub-Amazonian endemic regions (southern South America), while R. prolixus and T. dimidiata habit northern South America and Central America. Despite to the morbidity and mortality of CD, until now two old drugs are used to treat Chagas disease, namely the nitro derivatives based on imidazole and furan rings, – benznidazole (1) and nifurtimox (2), discovered in 1965 and in 1971, respectively. These drugs are known under the names Rochagan® and Lampit®, respectively (Figure 1).
Benznidazole and nifurtimox require long-term administration and often show efficacy problems, including insufficient activity in established, chronic forms of the disease (very low cure rates in the chronic stage). Having a bad reputation both in terms of safety and efficacy, these old medications must be replaced by new safer and more efficacious drugs. Some developed molecules are being investigated in clinical trials. Among them, it should be mentioned new 2-substituted 5-nitroimidazole, fexinidazole (3) and repurposed antifungal triazole, posaconazole (4) (Figure 1).
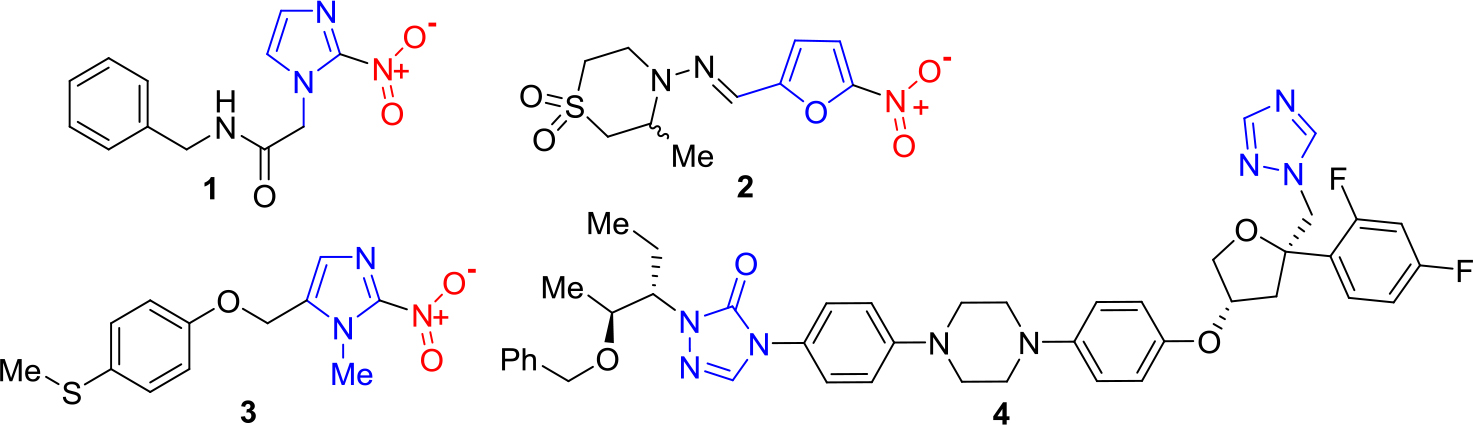
Figure 1. Chemical structures of anti-chagasic drugs and drug candidates based on azole scaffold.
Fexinidazole has been identified as a new promising drug candidate for treatment of African trypanosomiasis [6], but it was demonstrated that it could be used in the chemotherapy for Chagas disease [7, 8]. Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus was reported in 2010 [9].
Since this time, posaconazole is the drug with more advanced development in both experimental model and clinical trial [10, 11]. However, despite the promising results obtained in the animal model, the clinical trials of this drug candidate did not meet expectations. Testing a combination of posaconazole and beznidazole called chagasazol, it was concluded that this triazole is not effective as mono-pharmaceuticals in the treatment of patients in chronic asymptomatic phase with respect to beznidazole[12].
While the antiprotozoal toxicity of nitroimidazoles (1, 3) and nitrofurans (2) are thiol-dependent on reduction by nitroreductases of the nitro group, which generates cytotoxic species that cause damage to DNA, lipids and proteins [13], triazoles like posaconazole (4) are inhibitors of the sterol biosynthesis pathway, in particular C14-α-demethylase inhibitors [7, 14].
Thus, the approach to antiparasitic chemotherapy is usually based on two broad classes of targets, – trypanothione reductasa (TR) and sterol 14α-demethylase (CYP 51), which are specific to the parasite. Other alternative potential targets of responsible parasite for CD, T. cruzi are trans-sialidase (TS), tubulin, kinetoplastid DNA and cruzain (also referred to as cruzipain, the full-length native enzyme) [15, 16].
Cruzain and Identification of its Inhibitors
Cruzain (EC 3.4.22.51), the major papain-like cysteine peptidase of T. cruzi, is a principal virulence factor and a chemotherapeutic target with remarkable pre-clinical validation evidence [17–19].
The studies of the basic biochemistry of T. cruzi have allowed the identification of novel targets for CD chemotherapy. Killing T. cruzi through an inhibition of the enzyme cruzain is a different approach in therapies for Chagas disease. Being a member of the papain C1 family of cysteine protease, this enzyme hydrolyzes chromogenic peptides at the carboxyl arginine or lysine residue and requires at least one more amino acid between the terminal arginine or lysine and the amino-blocking group.
Thus, cruzain enzyme is essential for the viability and virulence of T. cruzi [20]. From currently available information in the Protein Data Bank (PDB) [21] it is known that cruzain consists of two domains: one with α-helices form and the other of antiparallel β-sheets. The active site, located at the interface between the two domains, encloses the catalytic triad formed by Cys25, His159 and Asn175. Cruzain has four relevant subsites for ligand binding (S1, S2, S3 and S1’) [22, 23].
During early development of cysteine protease inhibitors for CD therapy different class of many small peptidyl molecules were designed, synthetized and tested; among them, there were diazomethane inhibitors, fluoromethyl ketones, oxygen-containing heterocycles and vinyl sulfones. All these molecules bind a peptide or pseudopeptide backbone of cysteine protease (i.e., cruzain) to form a covalent adduct that inactivates the enzyme. Unfortunately, irreversible inhibitors may also inhibit host protease enzymes and possibly cause unacceptable side effects. Such irreversible inhibitors, especially peptidyl vinyl sulfones have become interesting leads in clinical trials [24].
The most outstanding vinyl sulfone, which was efficacious in pre-clinical models of T. cruzi infection, including immunocompetant and immunodeficient mice and dogs, is the irreversible cruzain inhibitor K-777(5) [25, 26]. Vinyl sulfone K-777 (5, known also as K11777), a potent cruzain inhibitor (IC50 = 5 nM) that inhibits the enzyme by attaching irreversibly to the catalytic Cys25 thiol group: first, conjugate addition of the thiolate of the cysteine at the active site to the double bond occurs that leads to the formation of carbanion, and then the resulting carbanion is subsequently protonated driving the process thermodynamically to the more stable enzyme−inhibitor complex (Figure 2).
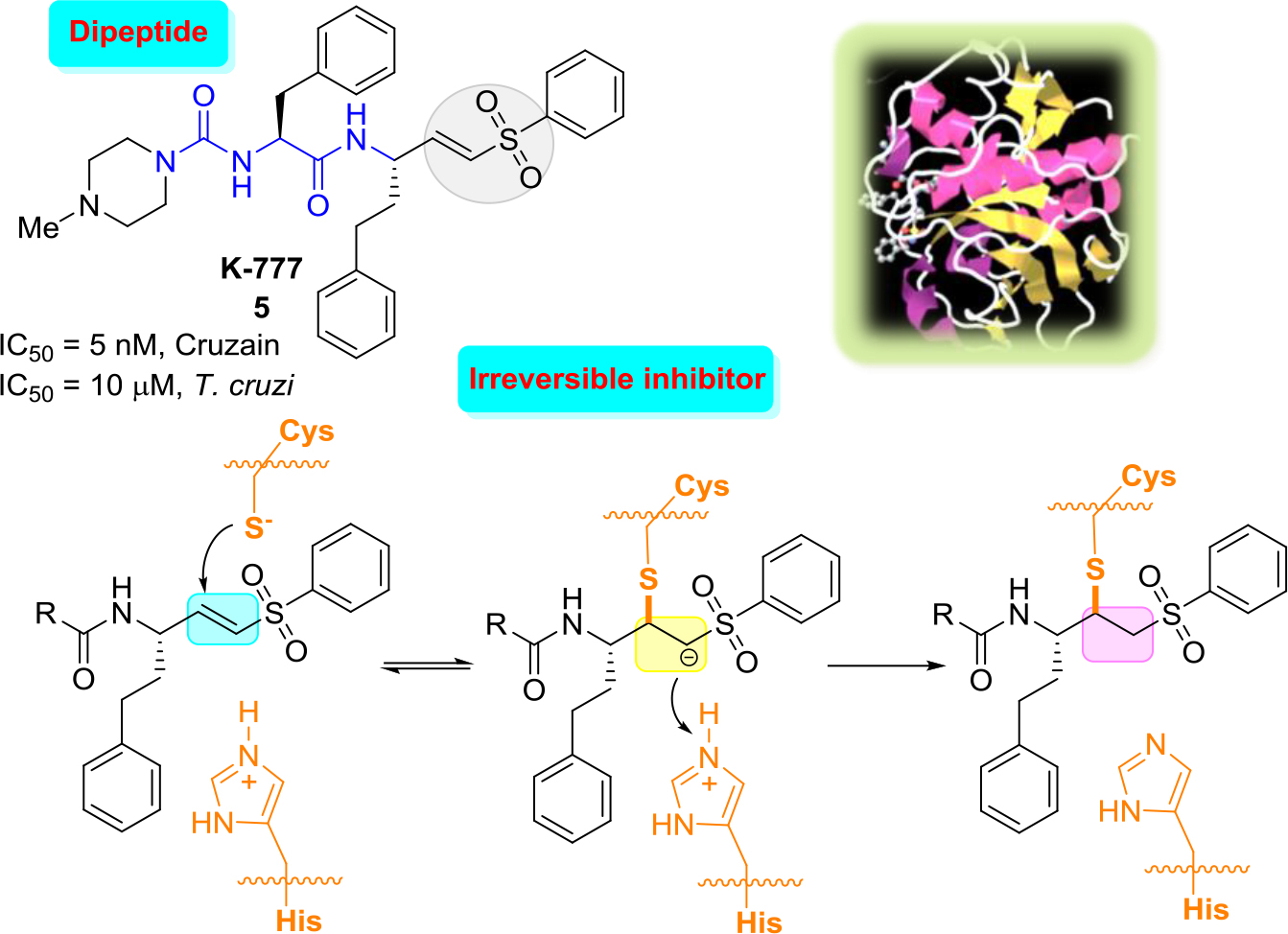
Figure 2. Structure of K-777, three-dimensional structural representation of cruzain crystallized with this inhibitor and simplified inhibition scheme.21
These results indicated that there were no laboratory abnormalities, histologic abnormalities, or auto-immune phenomena. However, in 2005 it was announced that due to severe problems related to elevated hepatotoxicity of K-777 could lead to the abandonment of this project [27]. Since then, a number of more rigorous follow-up safety studies were done. Thus, while K777 continues to undergo preclinical evaluation, which may lead towards a possible human clinical trial [28], more selective, reversible inhibitors would be more suitable, the ideal drugs.
In general, three are two approaches to designing reversible inhibitors: (a) modifying K777 structure and (b) modifying structure of active inhibitors of human cathepsins that were designed for other diseases related to these enzymes. It should be evoked that cruzain is closely related to the human cysteine protease family of cathepsins, especially cathepsins L, B and K, which is the primary enzyme involved in bone formation process.
Both routes have active development. Indeed, modifying K777 structure (approach a), recently diverse reversible inhibitors, which contain electrophilic groups that serve as a ‘warhead’ that binds to cruzipain through a covalent bond to the active site cysteine, have been designed. Among them were first epoxysulfones and then halogenated vinyl sulfones [29, 30] and dipeptidyl nitro alkenes [31]. The latter molecules like a nitro alkene (6) (Figure 3) resulted in a new type of highly potent covalent reversible inhibitors of cysteine proteases. The results showed that this dipeptidyl nitro alkene (6) should bind in a comparable manner as the corresponding vinyl sulfones. However, chemical nature of the arising nitroalkane carbanion is quite different to the one of agent K-777, since the acidity of the corresponding acid, namely, the nitro alkane, is higher, and thus, the basicity of the carbanion is lower [30]. Moreover, the authors commented that the docking experiments indicate that a reaction, i.e. a nucleophilic attack at the double bond, should be possible.
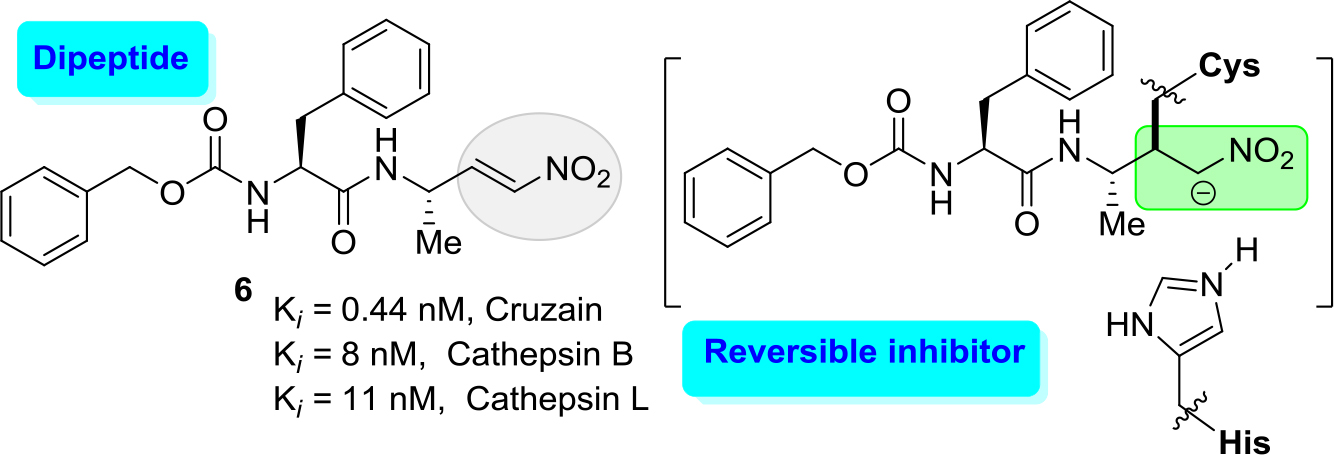
Figure 3. Potent inhibitor (6) of cruzain with Ki values of 0.44 nM.
However, it should be noted that this molecular inhibitor exhibited a high affinity for human cathepsins, cathepsin B and cathepsin L showing Ki values of 8 and 11 nM, respectively. Thus, this cruzain inhibitor lucks a good selectivity index (SI) of human cathepsins/cruzain ratio.
An excellent example of the approach (b) is the development of new cathepsin K inhibitors based on amidoacetonitrile moiety [23, 32–34] that led to the identification of a peptidomimetic α-amido nitrile MK-0822 called odanacatib (7) (Figure 4), which was selected in the clinical development for osteoporosis and bone metastasis in 2008 [35].
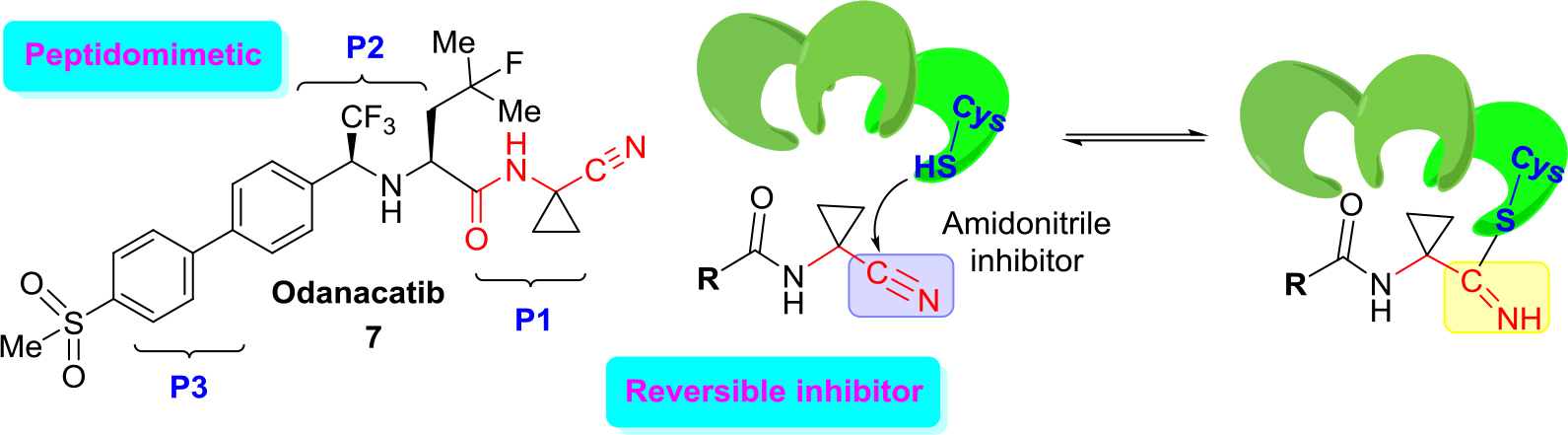
Figure 4. Structure of odanacatib and simplified scheme of its specific interaction with cruzain that occurs in the binding pocket with Cys-SH.
Unfortunately, in 2016 odanacatib was dropped because of adverse events [36]. However, specific interaction of this molecule and related aminonitrile compounds with cysteine proteases consists of the nucleophilic attack of the thiolate moiety (Cys-SH) at the triple C ≡ N bond and thus the reversible formation of thioimidate adducts suggested that reversible cruzain inhibitors could be successful anti-T. cruzi agents [37].
Given the structural similarities between cruzain and the cathepsins, a drug discovery effort was mounted to identify an odanacatib analogs locking for the best selectivity (human cathepsins/cruzain). Division of the scaffold of this drug into three portions that it was identified as P1, P2, and P3 aided much to find new suitable cruzain inhibitors. Realizing focused screening of diverse cysteine protease inhibitors collection, first it could be identified amidonitrile (8) with basic moieties in P3 portion of model drug odanacatib (7) [38].
However, the basic nature of (8) is likely associated with lysosomotropism in which the accumulation of the compound in lysosomes could lead to unexpected activity against human cathepsins. Diverse related studies confirmed the potential of cruzain inhibitors as anti-T. cruzi therapies and allowed to identify two orally active, reversible cruzain inhibitors Cz007 (9) and Cz008 (10) (Figure 5).
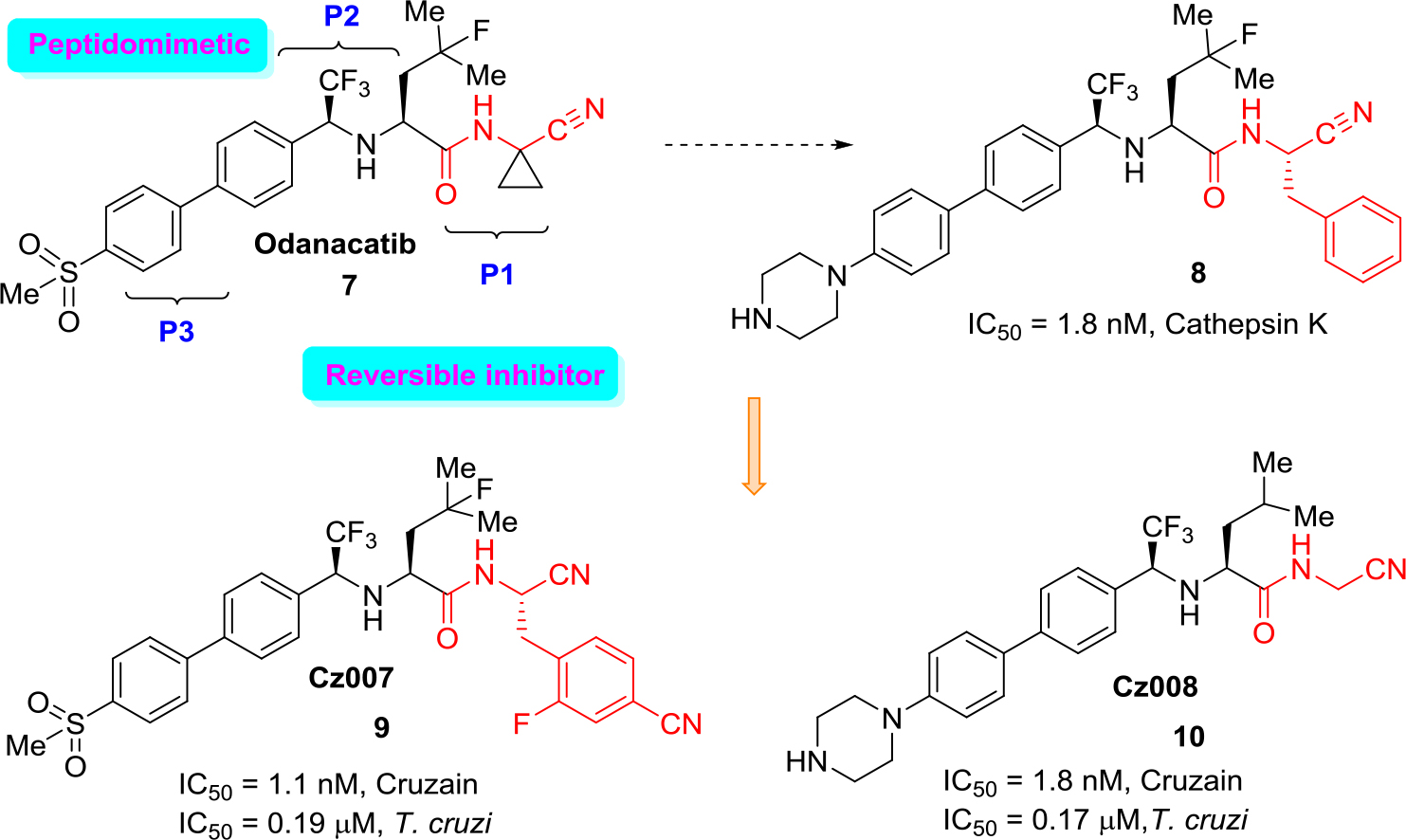
Figure 5. Drug odanacatib as a model for developing new reversible cruzain inhibitors.
These structural non peptidic amidonitrile analogs of odanacatib have been found to be potent cruzain inhibitors displaying in vivo activity in a murine model of acute T. cruzi infection [39, 40]. Recently, several large libraries of α-amidoacetonitriles were also prepared and tested their inhibitory activity against the T. cruzi cysteine protease cruzain and anti-trypanosomal effects. A Ki value of 16 nM and an EC50 value of 28 μM were observed for the most potent of these inhibitors [41].
In silico computational studies through Molinspiration software
Being small nitrogen-containing molecules, the above mentioned compounds (1–10) are capable to exert non-covalent interactions (hydrophobic and electrostatic interactions as well as hydrogen bonds, metal co-ordination, van der Waals forces, etc.) with some active sites in organisms that ends with pharmacological activity manifestation. This pharmacological activity is also influenced by certain simple physicochemical parameters (descriptors).
Usually, four most important parameters of small molecules are used to predict their oral bioavailability; these descriptors are molecular weight (MW), lipophilicity (Log P), H-bond donors (HBD) and H-bond acceptors (HBA). All these are involved in the Lipinski “rule of five” (RO5), which states that good absorption or permeation of a compound is more likely when there are less than 5 H-bond donors, 10 H-bond acceptors, the MW is smaller than 500 Da and the calculated Log P (cLog P) is smaller than 5 [42]. These simple filters can help in selecting drug-like compounds. However, RO5 compliance is not a guarantee that a compound will be drug-like. Additionally, other descriptors such as number of rotatable bonds (ROTB), topological polar surface area (TPSA) and molecular volume (MV) are considered very useful parameters in activity–structure relationship (SAR) studies. All these properties could be easily calculated by diverse software available online.
Considering above backgrounds and paying attention to the importance of knowing these physicochemical properties, we used Molinspiration software [43] for actual anti-chagastic drugs 1–4 and candidate drug, promising cruzain inhibitors 5–10 (Table 1).
Table 1. Molecular descriptors calculated for drugs and drug candidates (1–10) according to the Lipinski’s Rule using the Molinspiration Cheminformatics software.
|
Comp. |
MWa |
cLogPb |
cLogSc |
HBAd |
HBDe |
ROTBf |
TPSAg |
MVh |
n |
|
1 |
260.25 |
0.78 |
–2.618 |
7 |
1 |
5 |
92.75 |
224.99 |
0 |
|
2 |
287.30 |
0.71 |
–2.767 |
8 |
0 |
3 |
108.71 |
229.37 |
0 |
|
3 |
265.29 |
2.58 |
–3.783 |
6 |
0 |
4 |
72.88 |
220.23 |
0 |
|
4 |
762.86 |
6.15 |
–11.01 |
12 |
0 |
13 |
104.73 |
678.93 |
3 |
|
5 |
574.75 |
4.32 |
–6.975 |
8 |
2 |
11 |
98.81 |
531.04 |
1 |
|
6 |
383.45 |
3.81 |
–4.958 |
7 |
2 |
11 |
96.18 |
359.60 |
0 |
|
7 |
525.57 |
3.96 |
–6.847 |
6 |
2 |
10 |
99.06 |
439.67 |
1 |
|
8 |
595.68 |
5.85 |
–8.528 |
6 |
2 |
12 |
80.18 |
539.00 |
2 |
|
9 |
632.65 |
5.16 |
–8.876 |
7 |
2 |
12 |
122.85 |
527.23 |
2 |
|
10 |
487.57 |
4.33 |
–6.498 |
6 |
3 |
10 |
80.18 |
446.16 |
0 |
a Molecular Weight (g/mol); b Logarithm of the partition coefficient between n-octanol and water; c Logarithm of the solubility measured in mol/L; d Number of hydrogen-bond acceptors; e Number of hydrogen-bond donors; f Number of rotatable bonds; g Polar Surface Area (Å2); h Molecular volume.
Analyzing the results obtained, it can be noted that two classic anti-chagastic drugs, benznidazole (1) and nifurtimox (2), and drug candidate fexinidazole (3) are very small, highly soluble molecules with poor capacity of permeability through membranes (weak lipophilicity); its cLogP are 0.78, 0.71 and 2.58, respectively. In addition, they are molecules with low flexibility properties (ROTB = 3–5). In contrast, drug candidates (4–10) are more lipophilic molecules (6.15 <cLogP > 3.81) with elevated MW (383.45–762.86) and MV (359.60–678.93).
Among them, antifungal drug, posaconazole (4) that was proposed in CD treatment, resulted to be the biggest molecule with the highest lipophilicity parameter (cLogP = 6.15) and flexibility properties (ROTB = 13). Additionally, posaconazole has a better ability of intermolecular hydrogen bonding (HBA = 12), which is partially responsible for the interactions with secondary and tertiary structures of proteins and nucleic acids.
The physicochemical descriptors of cruzain inhibitor amidonitriles (8, 9) derived from drug odanacatib (7), are very close to the one of posaconazole that inhibits the ergosterol production by binding and inhibiting the lanosterol-14α-demethylase. Noteworthy, these cruzain inhibitors and posaconazole are structurally different, very lipophilic molecules with more than two RO5 violations.
Own odanacatib molecule, possessing MW more than 500 g/mol, has one RO5 violation. Moreover, its more elaborated amidonitrile derivative (10) does not show any RO5 violation like dipeptidyl nitro alkene (6), a K777 analog (Table 1). However, it should be commented that there are plenty of examples available for such prediction violation amongst the existing drugs [44].
Thus, both approaches to designing reversible inhibitors (modifying from drug odanacatib and agent K777) result in promising cruzain inhibitors as anti-T. cruzi therapies. However, it is believed that α-amidoacetonitriles derived from odanacatib have more outlooks in anti-chagastic drug research [45].
Interesting results were obtained also analyzing bioactivity score for these compounds. According to the predicted bioactivity score, drug and drug candidates (1–4) do not make to switch any tested biological targets, while especially designed cruzain inhibitor agents (5–10) may alter selectively proteases (Table 2) that confirm generally its biochemical behavior. Unfortunately, simple in silico evaluation of these drug candidates does not allow anticipating their SI properties.
Table 2. Molinspiration bioactivity score for compounds (1–10)*
|
Comp. |
GPCR ligand |
Ion channel modulator |
Kinase inhibitor |
Nuclear receptor ligand |
Protease inhibitor |
Enzyme inhibitor |
|
1 |
–0.33 |
–0.39 |
–0.49 |
–0.71 |
–0.05 |
–0.02 |
|
2 |
–0.93 |
–1.40 |
–0.73 |
–1.61 |
–0.81 |
–0.58 |
|
3 |
–0.47 |
–0.22 |
–0.38 |
–0.41 |
–0.38 |
–0.09 |
|
4 |
–1.45 |
–2.82 |
–2.34 |
–2.54 |
–1.12 |
–2.07 |
|
5 |
0.21 |
–0.20 |
–0.18 |
–0.17 |
0.65 |
0.07 |
|
6 |
0.36 |
0.10 |
–0.16 |
–0.19 |
0.45 |
0.11 |
|
7 |
0.34 |
0.01 |
–0.11 |
0.04 |
1.16 |
0.27 |
|
8 |
0.19 |
–0.40 |
–0.32 |
–0.25 |
0.74 |
–0.20 |
|
9 |
0.07 |
–0.65 |
–0.41 |
–0.22 |
0.81 |
–0.18 |
|
10 |
0.13 |
–0.11 |
–0.02 |
–0.12 |
0.58 |
–0.11 |
* Marked in bold parameters indicated a pronounced bioactivity score
Additionally, it can be noted that tested inhibitors (5–8) could modify other important biological targets: all four molecules would affect G protein–coupled receptor (GPCR) (score = 0.19–0.34), drug odanacatib (7) would work also as an enzyme inhibitor (score = 0.27). These protein covalent binding properties possibly cause unacceptable side effects.
Conclusion
Chagas disease represents an emerging worldwide challenge and there is an urgent, unmet need for safe and effective medication and thus, the last few years have seen a revolutionary improvement of the in vitro and in vivo screening methodologies to search for new anti-Chagas therapies.
The studies on reversible cruzain α-amidoacetonitrile-based inhibitors represent a valuable step in the search for new drugs for the treatment of a neglected disease that continues to affect the lives of millions of people.
The RO5 methodology appears to be as useful today in defining therapeutically relevant pharmacokinetic drugability and could help in this search. This simple and brief in silico analysis not only could help to design better new molecules with suitable pharmacological properties, but also comprehend its possible mechanism of action. It is evident that one of the important rational approaches for the fast advance of novel trypanocidal chemotherapy would be one focused on drugs ready to enter to clinical trials or on the clinical evaluation of drug association with existing trypanocidal agents to get extra effectiveness and fewer secondary effects [46].
References
- Rassi A Jr, Rassi A, Marin-Neto JA (2010) Chagas disease. Lancet 375: 1388–1402. [crossref]
- Bern C (2015) Chagas’ Disease. N Engl J Med 373: 456–466. [crossref]
- Cavalli A, Bolognesi ML (2009) Neglected tropical diseases: multi-target-directed ligands in the search for novel lead candidates against Trypanosoma and Leishmania. J Med Chem 52: 7339–7359.
- Chatelain E (2015) Chagas disease drug discovery: toward a new era. J Biomol Screen 20: 22–35. [crossref]
- Bonney KM (2014) Chagas disease in the 21st century: a public health success or an emerging threat? Parasite 21: 11. [crossref]
- Torreele E, Trunz BB, Tweats D, Kaiser M, Brun R, et al. (2010) Fexinidazole: a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl Trop Dis 4: 923.
- Caldas S, Caldas IS, Cecílio AB, Diniz LF, Talvani A, et al. (2014) Therapeutic responses to different anti-Trypanosoma cruzi drugs in experimental infection by benznidazole-resistant parasite stock. Parasitology 141: 1628–1637.
- Bahia MT, de Andrade IM, Martins TA, do Nascimento ÁF, Diniz Lde F, et al. (2012) Fexinidazole: a potential new drug candidate for Chagas disease. PLoS Negl Trop Dis 6: 1870. [crossref]
- Pinazo MJ, Espinosa G, Gállego M, López-Chejade PL, Urbina JA, et al. (2010) Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am J Trop Med Hyg 82: 583–587.
- Molina I, Salvador F, Sánchez-Montalvá A (2015) The use of posaconazole against Chagas disease. Curr Opin Infect Dis 28: 397–407.
- Molina I, Gómez i Prat J, Salvador F, Treviño B, Sulleiro E, et al. (2014) Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N Engl J Med 370: 1899–1908. [crossref]
- Alonso-Padilla J, Rodríguez A (2014) High throughput screening for anti-Trypanosoma cruzi drug discovery. PLoS Negl Trop Dis 8: 3259. [crossref]
- Hall BS (2011) Wilkinson SR. Activation of benznidazole by trypanosomal type I nitroreductases results in glyoxal formation. Antimicrob Agents Chemother 56: 115–123.
- Diniz LF, Urbina JA, de Andrade IM, Mazzetti AL, Martins TAF, et al. (2013) Benznidazole and posaconazole in experimental Chagas disease: positive interaction in concomitant and sequential treatments. PLoS Negl Trop Dis 7: 2367.
- Sueth-Santiago V, Decote-Ricardo D, Morrot A, Freire-de-Lima CG, Lima MEF (2017) Challenges in the chemotherapy of Chagas disease: Looking for possibilities related to the differences and similarities between the parasite and host. World J Biol Chem 8: 57–80.
- Lepesheva GI (2013) Design or screening of drugs for the treatment of Chagas disease: what shows the most promise? Expert Opin Drug Discov 8: 1479–1489. [crossref]
- Du X, Guo C, Hansell E, Doyle PS, Caffrey CR, et al. (2002) Synthesis and structure-activity relationship study of potent trypanocidal thio semicarbazone inhibitors of the trypanosomal cysteine protease cruzain. J Med Chem 45: 2695–2707.
- Cazzulo J, Stoka V, Turk V (2001) The major cysteine proteinase of Trypanosoma cruzi: a valid target for chemotherapy of Chagas disease. Curr Pharm Design 7: 1143–1156.
- Branquinha MH, Oliveira SSC, Sangenito LS, Sodre CL, Kneipp LF, det al. (2015) Cruzipain: an update on its potential as chemotherapy target against the human pathogen Trypanosoma cruzi. Curr Med Chem 22: 2225–2235.
- Duschak VG, Couto AS (2009) Cruzipain, the major cysteine protease of Trypanosoma cruzi: a sulfated glycoprotein antigen as relevant candidate for vaccine development and drug target. A review. Curr Med Chem 16: 3174–3202.
- Protein Data Bank, http://www.rcsb.org/pdb.
- Gillmor SA, Craik C, Fletterick RJ (1997) Structural determinants of specificity in the cysteine protease cruzain. Protein Sci 6: 1603–1611.
- Martinez-Mayorga K, Byler KG, Ramirez-Hernandez AI, Terrazas-Alvares DE (2015) Cruzain inhibitors: efforts made, current leads and a structural outlook of new hits. Drug Discov Today 20: 890–898.
- McKerrow JH (1999) Development of cysteine protease inhibitors as chemotherapy for parasitic diseases: insights on safety, target validation, and mechanism of action. Int J Parasitol 29: 833–837.
- Doyle PS, Zhou YM, Engel JC, McKerrow JH (2007) A cysteine protease inhibitor cures Chagas’ disease in an immunodeficient-mouse model of infection. Antimicrob Agents Chemother 51: 3932–3939.
- Barr SC, Warner KL, Kornreic BG, Piscitelli J, Wolfe A, et al. (2005) A cysteine protease inhibitor protects dogs from cardiac damage during infection by Trypanosoma cruzi. Antimicrob Agents Chemother 49: 5160–5161.
- Urbina JA (2010) Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Tropica 115: 55–68.
- Sajid M, Robertson SA, Brinen LS, McKerrow JH (2011) Cruzain: the path from target validation to the clinic. In Cysteine Proteases of Pathogenic Organisms. Robinson MW, and John P. Dalton JP, Eds. Adv Exp Med Biol. Boston: Springer 712: 100–115.
- Doherty W, James J, Evans P, Martin L, Adler N, et al. (2014) Preparation, anti-trypanosomal activity and localisation of a series of dipeptide-based vinyl sulfones. Org Biomol Chem 12: 7561–7571.
- Schirmeister T, Kesselring J, Jung S, Schneider T, Weickert A, et al. (2016) Quantum chemical-based protocol for the rational design of covalent inhibitors. J Am Chem Soc 138: 8332–8335.
- Latorre A, Schirmeister T, Kesselring J, Jung S, Johe´ P, et al. (2016) Dipeptidyl nitroalkenes as potent reversible inhibitors of cysteine proteases rhodesain and cruzain. ACS Med Chem Lett 7: 1073–1076.
- Ferreira LG, Andricopulo AD (2017) Targeting cysteine proteases in trypanosomatid disease drug discovery. Pharm Therap180: 49–61.
- Frizler M, Lohr F, Furtmann N, Kla¨s J, Gu¨tschow M (2010) Structural optimization of azadipeptide nitriles strongly increases association rates and allows the development of selective cathepsin inhibitors. J Med Chem 54: 396–400.
- Frizler M, Stirnberg M, Sisay MT, Gutschow M (2010) Development of nitrile-based peptidic inhibitors of cysteine cathepsins. Curr Top Med Chem 10: 294–322.
- Gauthier JY, Chauret N, Cromlish W, Desmarais S, Duong LT, et al. (2008) The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg Med Chem Lett 18: 923–928. [crossref]
- Mullard A (2016) Merck &Co. drops osteoporosis drug odanacatib. Nat Rev Drug Discov 15: 669. [crossref]
- Nicoll-Griffith DA (2012) Use of cysteine-reactive small molecules in drug discovery for trypanosomal disease. Expert Opin Drug Discov 7: 353–366.
- Black WC, Bayly CI, Davis DE, Desmarais S, Falgueyret JP, et al. (2005) Trifluoroethylamines as amide isosteres in inhibitors of cathepsin K. Bioorg Med Chem Lett 15: 4741–4744. [crossref]
- Beaulieu C, Isabel E, Fortier A, Masse F, Mellon C, et al. (2010) Identification of potent and reversible cruzipain inhibitors for the treatment of Chagas disease. Bioorg Med Chem Lett 20: 7444–7449.
- Ndao M, Beaulieu C, Black WC, Isabel E, Vasquez-Camargo F, et al. (2014) Reversible Cysteine Protease Inhibitors Show Promise for a Chagas Disease Cure. Antimicrob Agents Chemother 58: 1167–1178.
- Avelar LA, Camilo CD, de Albuquerque S, Fernandes WB, Gonçalez C, et al. (2015) Molecular Design, Synthesis and Trypanocidal Activity of Dipeptidyl Nitriles as Cruzain Inhibitors. PLoS Negl Trop Dis 9: 0003916.
- Lipinski CA (2004) Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 1: 337–341. [crossref]
- Molinspiration Cheminformatics Software, https://www.molinspiration.com .
- Benet LZ, Hosey CM, Ursu O, Oprea TI (2016) BDDCS, the Rule of 5 and drugability. Adv Drug Deliv Rev 101: 89–98. [crossref]
- Ang KK, Ratnam J, Gut J, Legac J, Hansell E, et al. (2011) Mining a cathepsin inhibitor library for new antiparasitic drug leads. PLoS Negl Trop Dis 5: e1023. [crossref]
- Duschak, VG (2016) Targets and patented drugs for chemotherapy of Chagas disease in the last 15 years-period. Recent Patentes Anti-infect Drug Discov 11: 74–173.