Abstract
Purpose: We recently demonstrated that offspring delivered to baboons deprived of estrogen during the second half of gestation exhibited insulin resistance. Therefore, because skeletal muscle accounts for >80% of insulin dependent glucose disposal, we suggested that estrogen in utero programs factors in fetal skeletal muscle important for insulin sensitivity in offspring. However, liver and adipose are also sites of insulin action and adipose insulin resistance can increase serum free fatty acid (FFA) levels and thereby reduce skeletal muscle insulin sensitivity. Therefore, in the current study we determined whether estrogen-deprived offspring exhibit normal adipose and hepatic function.
Results: The fasting serum levels of adiponectin, leptin, glucose, and analytes of liver function as well as the basal levels of serum FFA were similar in offspring of estrogen replete/suppressed baboons. Moreover, the normal glucose-induced decline in serum FFA levels measured in untreated offspring was also measured in offspring of letrozole-treated baboons. Fetal serum levels of adiponectin and leptin in late gestation also were similar and expression of nitrotyrosine negligible in fetal liver and adipose of untreated and letrozole-treated animals.
Conclusions: These results indicate that offspring of letrozole-treated baboons have normal adipose and liver function and do not exhibit adipose insulin resistance. Therefore, we suggest that the insulin resistance observed in estrogen-deprived offspring primarily reflects a decline in insulin-stimulated glucose clearance by skeletal muscle and which supports our original suggestion that estrogen in utero programs factors in fetal skeletal muscle that promote insulin sensitivity in offspring.
Key words
Estrogen, insulin sensitivity, offspring, primate, adipose
Introduction
It is well established that insulin resistance which is characterized as reduced sensitivity or responsiveness of target tissues, e.g. skeletal muscle, adipose and/or liver, to normal circulating levels of insulin, is a key feature of and typically precedes the onset of type 2 diabetes [1-3], a disease that is reaching epidemic levels in the United States and elsewhere [4,5]. However, our understanding of the factors that initiate or cause insulin resistance remains incomplete. We recently showed that offspring of baboon mothers in which maternal and fetal estradiol levels were suppressed >95% during the second half of gestation by maternal treatment with an aromatase inhibitor exhibited insulin resistance prior to and after onset of puberty [6,7]. Thus, the homeostasis model assessment of insulin resistance (HOMA-IR) was 2-fold higher and intra-venous glucose tolerance (iv GTT) impaired in offspring of letrozole-treated baboons compared to offspring exposed in utero to the normal increase in estradiol with advancing gestation. Impairment of insulin sensitivity has also been demonstrated in aromatase-null mice and in aromatase-lacking male and female human offspring that also developed in an estrogen-suppressed intrauterine environment [8-12]. Thus, we proposed that estradiol has a critical role in programming mechanisms within the developing fetus that lead to insulin sensitivity after birth [6].
In mammals including humans, skeletal muscle accounts for about 80% of total glucose disposal in response to insulin, and thus a reduction in whole-body response to insulin, i.e. insulin resistance, is thought to reflect a decline in insulin-stimulated glucose clearance by skeletal muscle [13]. Therefore, we have suggested that estrogen in utero programs factors in fetal skeletal muscle that promote insulin sensitivity in offspring [6]. In support of this suggestion, estradiol has been shown to stimulate insulin sensitivity and enhance glucose tolerance in adult skeletal muscle and mice lacking estrogen receptor (ER) α exhibit insulin resistance within skeletal muscle [14-16]. However, liver and adipose are also major sites of insulin action. Adipose resistance to insulin can lead to increased serum levels of free fatty acids (FFA), which can impair insulin signalling [17-19] and promote FFA oxidation and decrease glucose oxidation in muscle cells, leading to reduced glucose uptake and a progressive loss of insulin responsiveness in skeletal muscle [19]. Therefore, it is important to ascertain whether the insulin resistance elicited in offspring of baboons deprived of estrogen in utero reflects a concomitant alteration in adipose responsiveness to insulin.
It is well established that serum FFA levels steadily decline after a bolus of glucose, which reflects the normal response of adipose to the glucose-induced increase in insulin levels, a response which is significantly blunted in adipose resistance/type 2 diabetes [20,21]. Therefore, in the current study we measured the serum levels of FFA in offspring of untreated and letrozole-treated baboons before and after a bolus injection of glucose, that is during intravenous glucose tolerance test (ivGTT). Moreover, adipose insulin resistance is often associated with a marked decrease in the serum levels of adiponectin, which is produced by fat and promotes adipose function and protects adipose from factors (e.g. inflammation) that can impair adipose responsivity to insulin [22]. Therefore, we also measured the serum levels of adiponectin as well as leptin which is reflective of adiposity [23] in offspring of baboons untreated or treated with letrozole. Finally, to ascertain whether oxidative stress and/or levels of glycogen were also compromised by estrogen deprivation in utero, we measured the expression of nitrosylated protein in fetal adipose and fetal liver and levels of glycogen in fetal liver of baboons untreated or treated with letrozole.
Materials and Methods
Animals
Blood and tissue samples were obtained from animals studied previously as part of our ongoing studies of the role of estrogen on placental-fetal development [6, 7, 24, 25]. Briefly, female baboons (Papio anubis), originally obtained from the Southwest National Primate Research Center, San Antonio, TX, were housed individually in large primate cages in air-conditioned rooms with a 12h/12h light/dark lighting cycle and fed standard low-fat primate chow (Harlan Primate Diet, Madison, WI) twice daily, fresh fruit and vitamins daily and water ad libitum. Female baboons were paired with male baboons for 5 days at mid menstrual cycle and pregnancy confirmed by ultrasound. Pregnant baboons were then either untreated or treated daily between days 100 and 165-180 of gestation (term = 184 days) with the aromatase inhibitor letrozole (4,4-[1,2,3-triazol-1 yl-methylene]bis-benzonitrate, Novartis Pharma AG, Basel, Switzerland; 115 µg/kg body weight/day, via maternal sc injection in 1.0 ml sesame oil). Blood samples (2-3 ml) were obtained at 3-5 day intervals during the second half of gestation from a peripheral maternal saphenous vein after brief restraint and sedation with ketamine HCl (10 mg/kg body weight, im). On days 165-180 of gestation, fetuses were either delivered spontaneously or were anesthetized with isoflurane and after collection of blood samples (3-5 ml) from the umbilical vein and artery, were delivered by cesarean section to synchronize the timing of delivery. Some of the fetuses were immediately euthanized with pentobarbital (100 mg/kg body weight, iv) and adipose and liver tissue samples fixed in formaldehyde and embedded in paraffin or frozen and stored in liquid nitrogen.
For studies of postnatal development baboon newborns were left with and nursed by their mothers for 8 months at which time they were weaned and placed in pairs in cages adjacent to their respective mothers and fed standard low-fat primate chow (Harlan Primate Diet) twice daily, fresh fruit and vitamins daily and water ad libitum. Every 2-6 months thereafter baboon offspring were briefly sedated with ketamine HCl (10 mg/kg body weight, im), body weight measured, and blood samples (2-3 ml) obtained from a peripheral saphenous vein for the purpose of quantifying serum estradiol, adipokines, and analytes of hepatic and adipose function.
Intravenous glucose tolerance tests (ivGTT) were performed [6, 7] sequentially in baboon offspring between 4¼ – 8 years of age (i.e. postpuberty) and data averaged to yield a single value for each animal. Baboons were fasted overnight, sedated with ketamine HCl (2 mg/kg body weight, iv), administered a bolus of dextrose (0.25 gm/kg body weight) via an antecubital vein and blood samples (2.5 ml each) obtained from the saphenous vein before (0 time) and after dextrose administration. The serum samples obtained at 0, 5 and 30 minutes from baboon offspring in which the ivGTT was conducted at 6-8 years of age were used in the current study to examine adipose function as described below.
All baboons were cared for and used strictly in accordance with U.S. Department of Agriculture regulations and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th edition, National Academy Press, 2011). The Institutional Animal Care and Use Committees of the Eastern Virginia Medical School and the University of Maryland School of Medicine approved the experimental protocols employed in this study.
Assay of serum estradiol, adipokines, and analytes of hepatic function
Serum levels of estradiol in maternal saphenous vein during gestation and in umbilical vein at the time of delivery on days 165-180 were determined by radioimmunoassay using an automated chemiluminescent immunoassay system (Immulite; Siemens Healthcare Diagnostics, Deerfield, IL) as described previously [26]. The serum levels of leptin and total (low, medium and high molecular weight) adiponectin in umbilical artery at days 165-180 of gestation and in saphenous vein from offspring 6-8 years of age were determined using highly specific ELISA Sandwich Assay Kits and reagents and instructions supplied by the manufacturer (R&D Systems, Minneapolis, MN). The human leptin quantikine ELISA (HDLP 00) exhibited <0.5% cross reactivity with several factors/cytokines, intra- and inter-assay coefficients of variation of 3.0%-3.3% and 3.5%-5.4%, respectively, and a sensitivity of 7.8 pg/ml serum. The human adiponectin quantikine ELISA (Acrp 30) also exhibited <0.5% cross reactivity with several factors/cytokines and intra- and inter-assay coefficients of variation of 2.5%-4.7% and 5.8%-6.9%, respectively, and a sensitivity of 0.89 ng/ml serum. Adiponectin and leptin standards provided and baboon serum samples (10 µl diluted 1: 50 in reagent buffer) were assayed in duplicate and signal intensities for generation of standard curves which exhibited linearity were used for calculation of serum adipokine levels.
Levels of glucose, triglycerides, cholesterol, alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) in randomly-selected peripheral blood samples of 6-8 year old offspring were determined commercially (Antech Diagnostics, Lake Success, NY).
Analysis of glycogen content in fetal liver
The expression of glycogen in paraffin embedded sections (5 µm) of fetal liver was performed using the Best’s carmine procedure which stains glycogen red (Poly Scientific R&D Corp., Bay Shore, NY). The tissue levels of glycogen were determined using a Glycogen Assay Kit (Cayman # 700480) and reagents/instructions supplied by the manufacturer (Cayman Chemical, Ann Arbor, MI). Briefly, fetal liver tissue was homogenized, centrifuged at 800 x g and aliquots (in duplicate) and glycogen standards provided by the manufacturer incubated sequentially with amyloglucosidase to form β-D-glucose and then with glucose oxidase to yield D-glucono-δ-lactone and hydrogen peroxide. Following incubation with horseradish peroxidase the levels of the fluorescent product were measured fluorometrically at an excitation wavelength of 530-540 nm and an emission wavelength of 585-595 nm and glycogen levels expressed per mg tissue weight.
Western immunoblot of nitrotyrosine in fetal liver and adipose
Samples of fetal liver and perirenal and mesentery adipose were either homogenized (liver; 100 mg/ml) on ice in PBS containing 1% cholic acid, 0.1% SDS, 1 mM EDTA (Sigma-Aldrich, St Louis, MO) and a protease inhibitor cocktail, or polytroned in the same buffer (adipose; 250 mg/0.5 ml) containing 1% Igepal essentially as described previously [25]. Following centrifugation at 13,000 rpm for 30 minutes, samples (25 µg – 50 µg protein) and 2.5 µg and 5.0 µg of nitrotyrosine protein-enriched human liver homogenate standard (Millipore Sigma; ab 131380) were heated to 95C in Laemmli buffer, cooled, centrifuged (800 x gm), loaded onto 10% SDS-polyacrylamide gels (PAGE) and electrophoresed using a Bio-Rad Mini-Protean electrophoresis chamber (Bio-Rad Laboratories, Richmond, CA) in 25 mM Tris (pH 8.3), 192 mM glycine and 0.1% SDS. Proteins were wet-transferred onto an Immobilon-P membrane, blocked 1 h at room temperature with 3% non-fat dried milk in 10 mM Tris-HCl, pH 7.5, 150 M NaCl and 0.2% Tween 20 (TBST), and then incubated overnight (4C) with primary rabbit polyclonal antibody to nitrotyrosine (Millipore Sigma; 06-284) diluted 1: 1000 in TBST-1.5% non-fat dried milk, washed and then incubated for 1 h at room temperature with goat anti-rabbit horseradish peroxidase (HRP)-labeled secondary antibody (Vector Labs, Burlingame, CA) in TBST-1.5% non-fat dried milk. After washing in TBST, membranes were developed with enhanced chemiluminescence (GE Healthcare, Pittsburgh, PA) and exposed to Fuji Super RX medical x-ray film (Fujifilm Medical Systems, Roselle, IL). Membranes were then washed 3 times and incubated for 75 minutes at room temperature with primary mouse antibody to actin (Thermo Fisher, Waltham, MA; MAB1501) diluted 1: 5000 in TBST-1.5% non-fat dried milk, washed and then incubated for 1 h at room temperature with horse anti-mouse HRP-labeled secondary antibody (Vector Labs) in TBST-1.5% non-fat dried milk. After washing in TBST, membranes were developed and exposed to x-ray film as described above.
Assay of serum free fatty acid (FFA) in offspring during iv GTT
Serum FFA levels were determined using a coupled enzymatic fluorometric FFA Assay Kit (Cayman # 700310) and reagents/instructions supplied by the manufacturer (Cayman Chemical, Ann Arbor, MI). Serum samples (10 µl) from baboon offspring obtained immediately before and 5 and 30 minutes after bolus injection of dextrose and FFA standards were added to 96 well plates. Samples were then incubated with acyl CoA synthetase to catalyze fatty acid acylation of coenzyme A, which was oxidized by acyl CoA oxidase to generate hydrogen peroxide and then incubated with horseradish peroxidase. The levels of the fluorescent product were measured fluorometrically with excitation and emission wavelengths of 530-540 nm and 585-595 nm, respectively.
Statistical analysis
Data are expressed as means ± SE and were analyzed by the Student’s t test for independent observations or the Mann-Whitney “t” test when standard deviations between the groups were different or by ANOVA with replication using SAS statistical software (SAS Institutes).
Results
Serum estradiol levels
As shown previously [6, 7, 24, 25], maternal peripheral serum estradiol levels in untreated baboons rose from approximately 1.5 ng/ml on days 85-120 of gestation to approximately 4.0 ng/ml by day 175. Within 48-72 h of the onset of letrozole treatment on day 100, maternal serum estradiol levels decreased (P<0.001) to and remained at approximately 0.1-0.2 ng/ml. Serum umbilical vein estradiol concentrations (i.e. blood delivered to the fetus) of letrozole-treated baboons (0.04 ± 0.01 ng/ml) were also only 5% of that (P<0.001) in untreated animals (0.68 ± 0.26 ng/ml).
Fetal body and liver weights were similar in untreated and letrozole-treated animals (Table 1). The body weight of baboon offspring progressively increased (P<0.01) throughout postnatal life in a comparable manner and thus was similar at 5 years of age in animals untreated or treated in utero with letrozole (Table 1). Moreover, as shown previously [7] although basal fasting blood levels of glucose immediately prior to the glucose (dextrose) tolerance test were not different in offspring from baboons untreated or treated with letrozole, basal fasting insulin levels were approximately 2-fold greater (P=0.03) and thus the HOMA-IR was 2-fold greater (P<0.05) in offspring treated prenatally with letrozole (2.09 ± 0.47) than in untreated (1.11 ± 0.17) animals (Table 1).
Table 1. Fetal body and liver weight and body weight and HOMA-IR in postpubertal baboon offspring.
| Treatment | Fetus1 | Offspring2 | |||
| Body weight(gm) | Liver weight(gm) | Age(yrs) | Body weight(gm) | HOMA-IR | |
| Untreated | 828 ± 17 | 25.8 ± 1.9 | 5.4 ± 0.3 | 13.4 ± 0.8 | 1.11 ± 0.17 |
| Letrozole | 839 ± 32 | 26.1 ± 1.2 | 5.7 ± 0.3 | 14.3 ± 1.2 | 2.09 ± 0.47* |
Values are expressed as mean ± SE on the day of delivery (day 165-180) or on the day of iv glucose tolerance test in offspring from baboons untreated or treated on days 100-180 of gestation (term = 184 days) with letrozole (115 µg/kg body weight/day via maternal sc injection). N = 6-12/group.
1From Babischkin et al, 2016 (25)
2From Pepe et al, 2016 (7)
* P < 0.05 versus untreated (Student “t” Test)
As seen in Figure 1, the mean (± SE) peripheral fasting/basal serum levels of FFA prior to bolus injection of dextrose (0 time) were similar in 6-8 year old offspring delivered to baboons untreated (595 ± 57 µmoles/liter) or treated in utero with letrozole (506 ± 113 µmoles/liter). In untreated offspring, serum FFA levels declined (P <0.05) to values that were lower (P< 0.05) 5 minutes (415 ± 45 µmoles/liter) and 30 minutes (301 ± 46 µmoles/liter) after iv glucose injection. However, although letrozole-treated offspring exhibited insulin resistance, the serum FFA levels in these animals also declined (P <0.05) to values that were significantly lower (P<0.05) 5 minutes (332 ± 73 µmoles/liter) and 30 minutes (243 ± 48 µmoles/liter) after iv glucose and levels were not different from values in untreated baboons. Moreover, as seen in Figure 2, the respective serum levels of adiponectin and leptin in umbilical artery (i.e. fetus) on day 165-180 of gestation were similar in baboons untreated (14.6 ± 1.3 µg/ml; 5.8 ± 0.1 ng/ml, respectively) or treated with letrozole (18.0 ± 1.0 µg/ml; 5.7 ± 0.1 ng/ml) and remained similar in 6-8 year old offspring untreated (20.5 ± 0.2 µg/ml; 5.9 ± 0.1 ng/ml, respectively) or treated in utero with letrozole (20.2 ± 0.6 µg/ml; 5.8 ± 0.1 ng/ml, respectively).
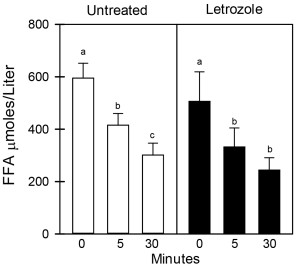
Figure 1. Serum levels of FFA before and 5 and 30 mins after iv glucose in 6-8 year old baboon offspring delivered to mothers untreated (n=6) or treated in utero with letrozole (n=6) as outlined in the legend to Table 1. Values (mean ± SE) with different letter superscripts differ from each other at P<0.05 (Repeated Measures ANOVA). Respective FFA levels before and after glucose challenge are not significantly different in untreated and letrozole-treated offspring (Student “t” tests).
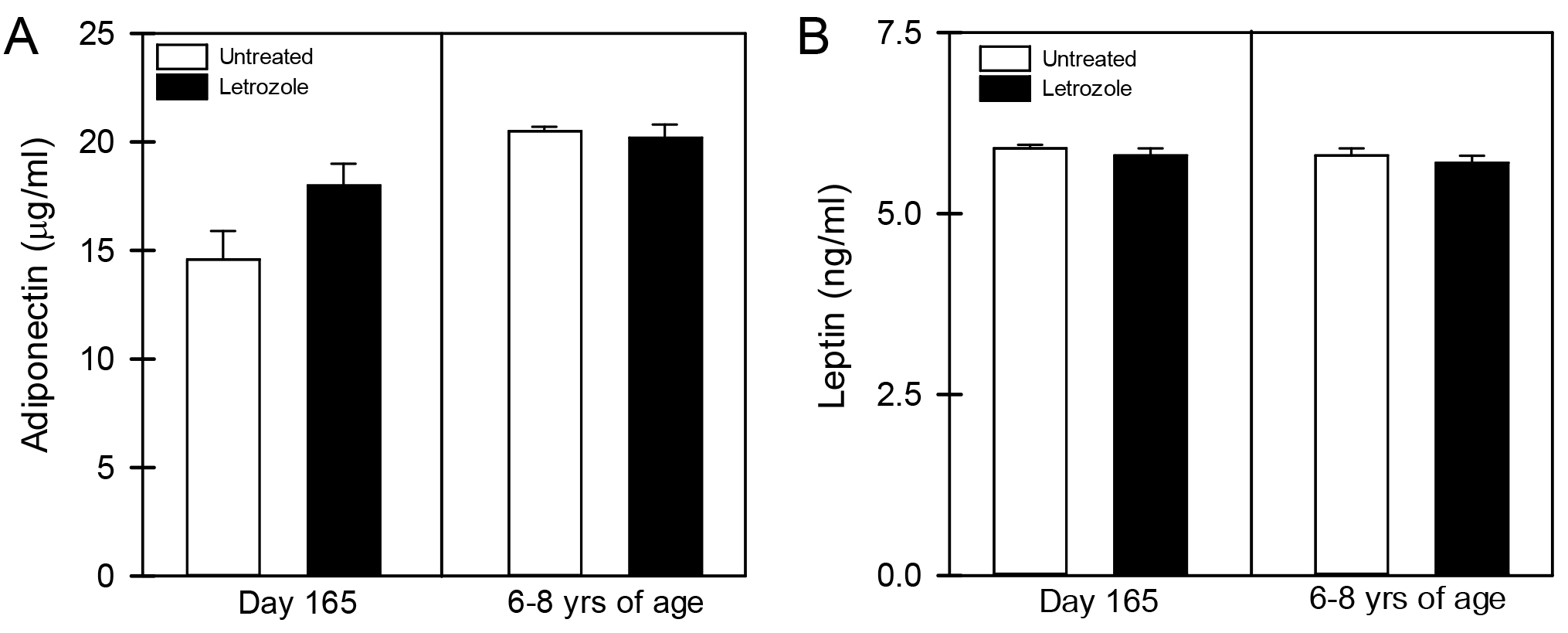
Figure 2. Mean (± SE) serum levels of adiponectin (A) and leptin (B) on day 165-180 of gestation in umbilical artery of fetuses and in saphenous vein of offspring 6-8 years of age born to mothers untreated or treated in utero with letrozole (n = 6-11/group).
Table 2. Levels of blood glucose, serum triglycerides, cholesterol and analytes of hepatic function in postpubertal baboon offspring1
| Treatment | Glucose (mg/dl) | Triglycerides (mg/dl) | Cholesterol (mg/dl) | AST (IU/L) | ALT (IU/L) | ALP (IU/L) |
| Untreated | 72 ± 4 | 55 ± 4 | 109 ± 7 | 8.1 ± 2.0 | 5.0 ± 0.0 | 152 ± 9 |
| Letrozole | 69 ± 5 | 56 ± 7 | 113 ± 8 | 5.8 ± 0.7 | 5.1 ± 0.1 | 139 ± 7 |
1 Values (mean ± SE) determined in peripheral blood samples obtained after overnight fast from 6-8 year old baboon offspring born to animals untreated (n = 8) or treated during the second half of gestation with letrozole (n = 6) as described in legend to Table 1. AST = aspartate aminotransferase; ALT =alanine aminotransferase; ALP = alkaline phosphatase.
As seen in Table 2, the respective fasting levels of glucose, triglycerides and cholesterol, as well as analytes of hepatic function including AST, ALT and ALK, were similar in blood samples obtained from 6-8 year old offspring delivered to baboons untreated or treated with letrozole.
In contrast to the abundant expression of nitrotyrosine protein in the standard nitrotyrosine protein enriched human liver homogenate provided by the manufacturer, nitrotyrosine was not detected in protein extracted from fetal perirenal or mesenteric adipose or fetal liver of baboon offspring untreated or treated in utero with letrozole and in which expression of actin was abundant (Figure 3).
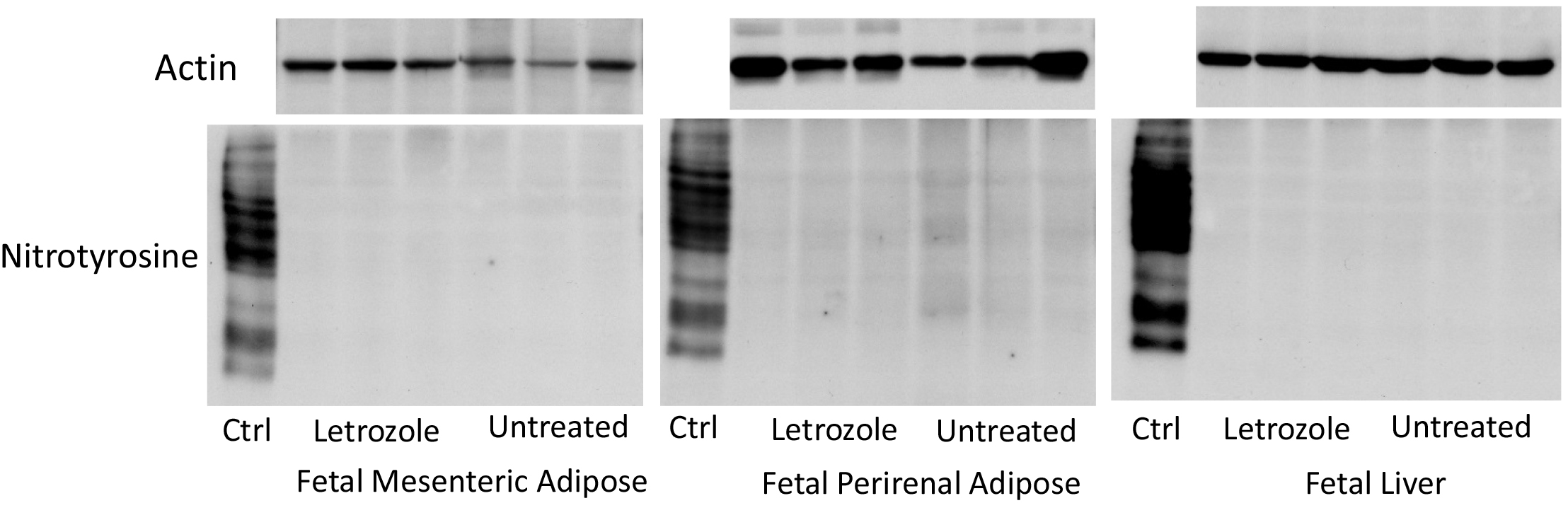
Figure 3. Representative immunoblots of nitrotyrosine protein and actin expression in extracts of fetal mesenteric and perirenal adipose and fetal liver obtained on days 165-180 of gestation from baboons untreated (n = 6) or treated in utero with letrozole (n = 6). Ctrl: expression of nitrotyrosine in nitrotyrosine protein enriched human liver homogenate provided by the manufacturer.
The levels of glycogen in fetal liver on days 165-180 of gestation were similar in untreated (142 ± 87 µg/mg tissue) and letrozole-treated (233 ± 72 µg/mg tissue) baboons (Figure 4).
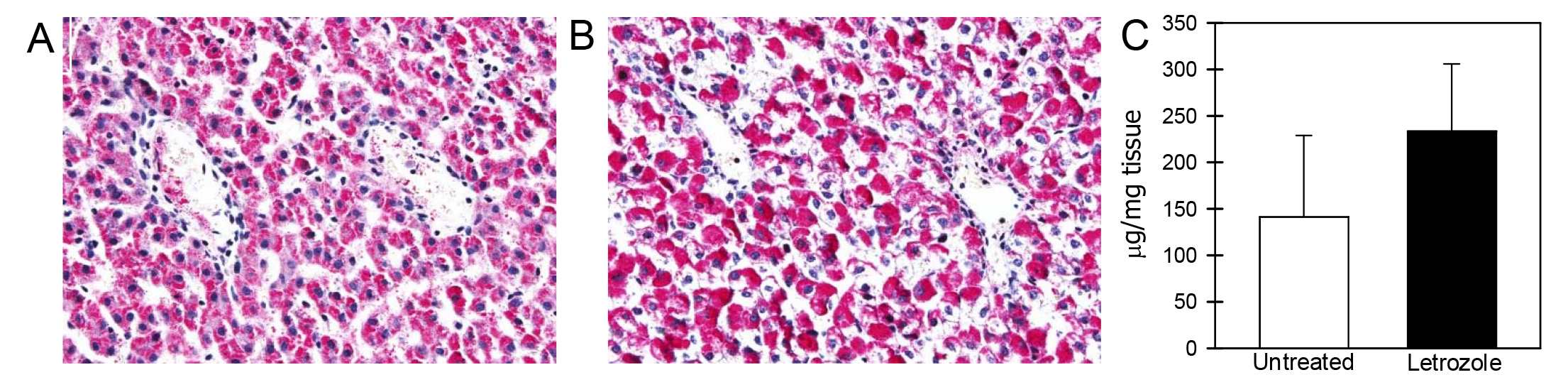
Figure 4. Glycogen expression (pink/red) in fetal liver on day 165 of gestation in baboons untreated (A) or treated with letrozole (B). Original magnification in Panels A, B = 200x. Panel C: Mean (± SE) concentrations (µg/mg tissue) of glycogen in fetal liver on days 165-180 of gestation in baboons untreated (n = 5) or treated with letrozole (n = 5).
Discussion
The results of the current study show that although offspring deprived of estrogen in utero exhibit insulin resistance [6,7], the basal and glucose-induced decline in serum levels of FFA in these animals were comparable to values in offspring exposed to the normal increase in estrogen with advancing gestation. Multiple studies show that the steady decline in serum FFA levels after an iv injection of glucose reflects the normal response of adipose to increased levels of insulin [20,21] which induces adipose lipoprotein lipase enzyme activity and thus a reduction in formation and release of FFA into blood [19]. Therefore, we suggest that offspring of letrozole-treated baboons do not exhibit adipose resistance to insulin. In support of this suggestion, serum levels of adiponectin and leptin in 6-8 year old offspring as well as fetuses near term were similar in baboons untreated or treated in utero with letrozole. Adiponectin promotes adipose function and protects adipose from factors (e.g. inflammation) that can impair adipose responsivity to insulin [22]. Thus, adipose insulin resistance is typically associated with and consequent to a decrease in the serum levels of adiponectin [21,22]. Leptin, also produced by adipose conveys information to the brain regarding the size of energy stores e.g. adipose, and can modulate neural centers that regulate intake of substrate and expenditure of energy [23,27]. Accordingly, adipose expression and serum levels of leptin increase exponentially with increasing fat mass and are increased in obese individuals [23,28,29]. Thus, the similar low levels of leptin in untreated and estrogen-deprived baboon offspring of the current study presumably reflects the comparable body weights in these non-obese animals fed a relatively low fat diet. In addition, we have previously shown that serum levels of a panel of inflammatory cytokines, which are often produced in excess by adipose in obese individuals, were also similar in offspring untreated or treated in utero with letrozole [6,7]. Collectively, these findings are highly supportive of our suggestion that estrogen-deprived offspring do not exhibit adipose resistance to insulin at this point in their development.
The current study also showed that fasting levels of glucose measured at the time of glucose tolerance testing and at other times during development in 6-8 year old offspring and hepatic glycogen levels in fetuses were similar in baboons untreated or treated with letrozole. An increased fasting level of blood glucose is typically observed in glucose intolerant individuals due to impaired insulin action in liver and which results in decreased glycogen synthesis and increased hepatic secretion of glucose in part due to increased activity of glucose-6-phosphatase [13, 21]. Moreover, several measures of liver function including hepatic enzymes as well as serum triglycerides and cholesterol were also comparable in offspring untreated or deprived of estrogen in utero. Therefore, it appears that hepatic metabolic function of offspring born to estrogen-suppressed baboons is normal and that the liver is not insulin resistant, although additional studies e.g. hyperinsulinemic-euglycemic clamp [30, 31], are required to establish the latter.
We propose, based on our previous findings [6, 7] and the results of the current study, that the insulin resistance elicited in offspring of baboons deprived of estrogen in utero primarily reflects a decline in insulin-stimulated glucose clearance by skeletal muscle. The latter is consistent with previous suggestions of others that insulin resistance reflects a decline in insulin-stimulated glucose clearance by skeletal muscle, simply because skeletal muscle, accounts for over 80% of total glucose disposal in response to insulin [13]. Moreover, since the basal and insulin-stimulated FFA levels were comparable in offspring of estrogen replete and estrogen suppressed baboons the proposed insulin resistance in skeletal muscle of offspring from estrogen-suppressed baboons is not due to elevated levels of FFA which promote FFA oxidation and decrease glucose oxidation in muscle leading to reduced glucose uptake and decreased insulin sensitivity in skeletal muscle [19]. Thus, these findings support our original suggestion that estrogen in utero programs factors in fetal skeletal muscle that promote insulin sensitivity in offspring [6].
Although offspring of letrozole-treated baboons do not exhibit adipose or hepatic insulin resistance, they have developed a deficit in first-phase pancreatic insulin release [7]. The latter coupled with skeletal muscle insulin resistance is often a prelude to development of type 2 diabetes, which would ostensibly ultimately include insulin dysfunction in adipose and liver [1-3]. It is important to point out that baboon offspring in our primate colonies are fed a diet rich in protein and low in fat and monosaccharides and are not obese. Thus, it is possible that if the letrozole-treated offspring were fed a high fat diet with/without high monosaccharides to mimic what is typically known as the “Western” diet to induce obesity the latter coupled with existing insulin resistance and first-phase pancreatic dysfunction, would likely rapidly progress to type 2 diabetes. Although the latter remains to be determined, it has been shown that otherwise normal baboons fed a high fat diet enriched with monosaccharides for 8 weeks developed aspects of cardiometabolic syndrome including adiposity, decreased adiponectin and insulin resistance [32].
It is well established that in pregnancies complicated by IUGR associated with hypoxia or poor fetal growth elicited by preterm birth in humans [33-35] and nonhuman primates [36-38] or by experimental manipulation of uteroplacental blood flow [39] or maternal nutrient restriction [40, 41], offspring are at high risk for development of insulin resistance which often progresses to type 2 diabetes [42-45]. However, as previously shown [6, 7], placental and fetal weights, maternal glucose tolerance/insulin action and uterine artery and umbilical blood flow [46] were not decreased in our letrozole-treated/estrogen-deprived baboons. Moreover, as shown in the current study, fetal perirenal and mesentery adipose and fetal liver of letrozole-treated animals did not exhibit oxidative/nitrosylated stress. Therefore, the action of estrogen in utero in programming fetal skeletal muscle insulin sensitivity/glucose homeostasis in adulthood appears to be specific and does not reflect alterations in maternal physiology as we have previously suggested [6]. This is in contrast to the effects of excess androgen administration to monkeys which induces insulin resistance in offspring, but also alterations in maternal parameters most notably increased maternal weight gain and glucose intolerance [47].
In summary, the results of the current study show that in baboon offspring deprived of estrogen in utero, fasting levels of adiponectin, leptin and glucose and basal serum levels of and the glucose-induced decline in serum FFA levels were similar to respective values in offspring exposed to the normal increase in estrogen with advancing gestation. Moreover, fetal serum umbilical artery levels of adiponectin and leptin were similar and expression of nitrotyrosine protein negligible in fetal liver and adipose in untreated and letrozole-treated fetuses. Therefore, we suggest that offspring of letrozole-treated baboons do not exhibit hepatic or adipose resistance to insulin and that the insulin resistance and the development of a deficit in first-phase pancreatic insulin release in these estrogen-deprived offspring primarily reflects a decline in insulin-stimulated glucose clearance by skeletal muscle. Collectively, these findings support our original suggestion that estrogen in utero programs factors in fetal skeletal muscle that promote insulin sensitivity in offspring [6].
Acknowledgments: This research was supported by National Institutes of Health Research Grant R01 DK 93950. The secretarial assistance of Ms. Sandra Huband with preparation of the figures and computer word processing of the manuscript and the assistance of Ms. Marcia Burch and Mr. Jeff Babischkin with the Western blots are sincerely appreciated. We thank Novartis Pharma (Basel, Switzerland) for generously providing the aromatase inhibitor letrozole to conduct this study.
Disclosure: The authors have nothing to disclose.
Conflict of interest: The authors declare that they have no conflict of interest.
References
- DeFronzo RA, Bonadonna RC, Ferrannini E (1992) Pathogenesis of NIDDM. A balanced overview. Diabetes Care 15: 318–368. [crossref]
- Lillioja S, Mott DM, Howard BV, Bennett PH, Yki-Jarvinen H, et al. (1988) Impaired glucose tolerance as a disorder of insulin action. Longitudinal and cross-sectional studies in Pima Indians. N Engl J Med 318: 1217–1225.
- Del Prato S, Tiengo A (2001) The importance of first-phase insulin secretion: implications for the therapy of type 2 diabetes mellitus. Diabetes Metab Res Rev 17: 164–174.
- Beltrán-Sánchez H, Harhay MO, Harhay MM, McElligott S (2013) Prevalence and trends of metabolic syndrome in the adult U.S. population, 1999–2010. J Am Coll Cardiol 62: 697–703. [crossref]
- Patra J, Jha P, Rehm J, Suraweera W (2014) Tobacco smoking, alcohol drinking, diabetes, low body mass index and the risk of self-reported symptoms of active tuberculosis: individual participant data (IPD) meta-analyses of 72,684 individuals in 14 high tuberculosis burden countries. PLoS One 9: e96433. [crossref]
- Maniu A, Aberdeen GW, Lynch TJ, Nadler JL, Kim SO, et al. (2016) Estrogen deprivation in primate pregnancy leads to insulin resistance in offspring. J Endocrinol 230: 171–183.
- Pepe GJ, Maniu A, Aberdeen G, Lynch TJ, Kim SO (2016) Insulin resistance elicited in postpubertal primate offspring deprived of estrogen in utero. Endocrine 54: 788–797.
- Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, et al. (2000) Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity. Proc Natl Acad Sci U S A 97: 12735–12740. [crossref]
- Takeda K, Toda K, Saibara T, Nakagawa M, Saika K, et al. (2003) Progressive development of insulin resistance phenotype in male mice with complete aromatase (CYP19) deficiency. J Endocrinol 176: 237–246.
- Belgorosky A, Guercio G, Pepe C, Saraco N, Rivarola MA (2009) Genetic and clinical spectrum of aromatase deficiency in infancy, childhood and adolescence. Horm Res 72: 321–330. [crossref]
- Remacle C, Dumortier O, Bol V, Goosse K, Romanus P, et al. (2007) Intrauterine programming of the endocrine pancreas. Diabetes Obes Metab 9 Suppl. 2: 196–209.
- Mitanchez-Mokhtari D, Lahlou N, Kieffer F, Magny J.F, Roger M, et al. (2004) Both relative insulin resistance and defective islet beta-cell processing of proinsulin are responsible for transient hyperglycemia in extremely preterm infants. Pediatrics 113: 537–541.
- DeFronzo RA, Gunnarsson R, Björkman O, Olsson M, Wahren J (1985) Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest 76: 149–55
- Margolis KL, Bonds DE, Rodabough RJ, Tinker L, Phillips LS, et al. (2004) Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women’s Health Initiative Hormone Trial. Diabetologia
47: 1175–1187. - Karjalainen A, Paassilta M, Heikkinen J, Backstrom AC, Savolainen M, Kesaniemi YA (2001) Effects of peroral and transdermal oestrogen replacement therapy on glucose and insulin metabolism. Clin. Endocrinol (Oxf) 54: 165–173.
- Manrique C, Lastra G, Habibi J, Mugerfeld I, Garro M, et al. (2012) Loss of Estrogen Receptor α Signaling Leads to Insulin Resistance and Obesity in Young and Adult Female Mice. Cardiorenal Med 2: 200–210. [crossref]
- Yu C, Chen Y, Cline G.W, Zhang D, Zong H, et al. (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 277: 50230–50236.
- Kruszynska YT, Worrall DS, Ofrecio J, Frias JP, Macaraeg G, et al. (2002) Fatty acid-induced insulin resistance: decreased muscle PI3K activation but unchanged Akt phosphorylation. J Clin Endocrinol Metab 87: 226–234.
- Ruan H, Lodish HF (2003) Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev 14: 447–455. [crossref]
- Prando R, Cordera R, De M.A, Maiello M, Odetti P, et al. (1978) I.V. glucose tolerance test: correlation between FFA, glucose and IRI in normal, obese and diabetic subjects. Acta Diabetol Lat 15: 259–272.
- Soriguer F, Garcia-Serrano S, Garcia-Almeida J.M, Garrido-Sanchez L, et al. (2009) Changes in the serum composition of free-fatty acids during an intravenous glucose tolerance test. Obesity (Silver Spring) 17: 10–15.
- Kishida K, Funahashi T, Shimomura I (2014) Adiponectin as a routine clinical biomarker. Best Pract Res Clin Endocrinol Metab 28: 119–130. [crossref]
- Mantzoros CS, Moschos SJ (1998) Leptin: in search of role(s) in human physiology and pathophysiology. Clin Endocrinol (Oxf) 49: 551–567. [crossref]
- Pepe G.J, Maniu A, Aberdeen G, Lynch T.J, Albrecht E.D (2016) Estrogen regulation of fetal adrenal cortical zone-specific development in the nonhuman primate impacts adrenal production of androgen and cortisol and response to ACTH in females in adulthood. Endocrinology 157: 1905–1913.
- Babischkin J.S, Aberdeen G.W, Pepe G.J, Albrecht E.D (2016) Estrogen suppresses interaction of melanocortin 2 receptor and its accessory protein in the primate fetal adrenal cortex. Endocrinology 157, 4588–4601.
- Albrecht ED, Aberdeen GW, Pepe GJ (2000) The role of estrogen in the maintenance of primate pregnancy. Am J Obstet Gynecol 182: 432–438. [crossref]
- Flier JS1 (1997) Leptin expression and action: new experimental paradigms. Proc Natl Acad Sci U S A 94: 4242–4245. [crossref]
- Lönnqvist F, Nordfors L, Jansson M, Thörne A, Schalling M, et al. (1997) Leptin secretion from adipose tissue in women. Relationship to plasma levels and gene expression. J Clin Invest 99: 2398–2404. [crossref]
- Sørensen TI, Echwald S, Holm JC (1996) Leptin in obesity. BMJ 313: 953–954. [crossref]
- Finegood D.T, Bergman R.N, Vranic M (1987) Estimation of endogenous glucose production during hyperinsulinemic-euglycemic glucose clamps. Comparison of unlabeled and labeled exogenous glucose infusates. Diabetes 36: 914–924.
- Muniyappa R, Lee S, Chen H, Quon M.J (2008) Current approaches for assessing insulin sensitivity and resistance in vivo: advantages, limitations, and appropriate usage. Am J Physiol Endocrinol Metab 294, E15–E26.
- Higgins P.B, Bastarrachea R.A, Lopez-Alvarenga J.C, Garcia-Forey M, Proffitt J.M (2010) Eight week exposure to a high sugar high fat diet results in adiposity gain and alterations in metabolic biomarkers in baboons (Papio hamadryas). Cardiovasc. Diabetol 9: 71.
- Barker DJ (2005) The developmental origins of insulin resistance. Horm Res 64: 2–7.
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL (2008) Effect of in utero and early-life conditions on adult health and disease. N Engl J Med 359: 61–73. [crossref]
- Gatford KL, Simmons RA, De Blasio MJ, Robinson JS, Owens JA (2010) Review: Placental programming of postnatal diabetes and impaired insulin action after IUGR. Placenta 31: Suppl, S60–S65.
- Blanco CL, Liang H, Joya-Galeana J, DeFronzo RA, McCurnin D, et al. (2010) The ontogeny of insulin signaling in the preterm baboon model. Endocrinology 151: 1990–1997. [crossref]
- Blanco CL, McGill-Vargas LL, Gastaldelli A, Seidner SR, McCurnin D.C, et al. (2015) Peripheral insulin resistance and impaired insulin signaling contribute to abnormal glucose metabolism in preterm baboons. Endocrinology 156: 813–823.
- McGill-Vargas L, Gastaldelli A, Liang H, Anzueto G.D, Johnson-Pais T, et al. (2017) Hepatic insulin resistance and altered gluconeogenic pathway in premature baboons. Endocrinology 158: 1140–1151.
- Simmons RA, Templeton LJ, Gertz SJ (2001) Intrauterine growth retardation leads to the development of type 2 diabetes in the rat. Diabetes 50: 2279–2286. [crossref]
- Ozanne SE, Smith GD, Tikerpae J, Hales CN (1996) Altered regulation of hepatic glucose output in the male offspring of protein-malnourished rat dams. Am J Physiol 270: E559–E564.
- Choi J, Li C, McDonald TJ, Comuzzie A, Mattern V, et al. (2011) Emergence of insulin resistance in juvenile baboon offspring of mothers exposed to moderate maternal nutrient reduction. Am J Physiol Regul Integr Comp Physiol 301: R757–R762
- Beltrand J, Lévy-Marchal C (2008) Pathophysiology of insulin resistance in subjects born small for gestational age Best Pract Res Clin Endocrinol Metab 22: 503–515. [
- Dunlop K, Cedrone M, Staples JF, Regnault T R (2015) Altered fetal skeletal muscle nutrient metabolism following an adverse in utero environment and the modulation of later life insulin sensitivity Nutrients 7: 1202–1216.
- Nijland MJ, Mitsuya K, Li C, Ford S, McDonald TJ, et al. (2010) Epigenetic modification of fetal baboon hepatic phosphoenolpyruvate carboxykinase following exposure to moderately reduced nutrient availability. J Physiol 588: 1349–1359.
- Li C, Levitz M, Hubbard GB, Jenkins SL, Han V, et al. (2007) The IGF axis in baboon pregnancy: Placental and systemic responses to feeding 70% global ad libitum diet Placenta 28: 1200–1210.
- Aberdeen G W, Baschat A A, Harman C R, Weiner C P, Langenberg P W, et al. (2010) Uterine and fetal blood flow indexes and fetal growth assessment after chronic estrogen suppression in the second half of baboon pregnancy. Am J Physiol Heart Circ Physiol 298: H881–H889.
- Abbott DH, Bruns CR, Barnett DK, Dunaif A, Goodfriend, et al. (2010) Experimentally induced gestational androgen excess disrupts glucoregulation in rhesus monkey dams and their female offspring Am J Physiol Endocrinol Metab 299: E741– E751.