Abstract
Triple-negative breast cancers (TNBC) lack estrogen receptor (ER), progesterone receptor and epidermal growth factor receptor HER2 and share many features of basal-like breast cancer (BLBC). Therefore, these patients have to rely on chemotherapy. The mechanistic target of rapamycin complex 1 (mTORC1) as well as its downstream targets p70 S6 kinase (S6K1) and S6K2 have been implicated in breast cancer. The 40S ribosomal protein S6 kinase 2 (S6K2) has been associated with endocrine resistance. We have previously shown that S6K2 protects against apoptotic cell death in ER-positive breast cancer cells. In the present study, we investigated if S6K2 could serve as a potential target for TNBC. Our analysis of TCGA dataset as well as immunohistochemistry of patient samples revealed that S6K2 is overexpressed not only in ER-positive but also in TN breast tumors compared to normal breast tissues. Silencing of S6K2 by siRNA enhanced sensitivity of BLBC MCF10CA1d cells to chemotherapeutic drugs cisplatin and doxorubicin. S6K2 knockdown also increased sensitivity of BLBC MCF10CA1a and TNBC HCC1395 cells to TRAIL. While S6K2 knockdown alone had little effect on apoptosis, it enhanced TRAIL-induced apoptosis as judged by the increase in caspase-3 activity, PARP cleavage and annexin V/PI staining. Overexpression of constitutively-active S6K2 construct in MDA-MB-231 cells protected against TRAIL-induced apoptosis. These results suggest that S6K2 also promotes survival of TNBC. Therefore, targeting S6K2 in combination with chemotherapeutic agents could improve therapy of TNBC.
Keywords
Triple-negative breast cancer, basal-like breast cancer, S6K2, chemotherapeutic drugs, apoptosis
Introduction
Breast cancer is the second leading cause of cancer-related death among women in the United States. The major breast cancer subtypes include hormone receptor-positive, human epidermal growth factor receptor 2 (HER2)-enriched and triple-negative or basal-like breast cancer [1]. Triple-negative breast cancers (TNBC), which account for approximately 20% of all breast cancers, are a highly aggressive form of breast cancer with high mortality and poor prognosis [2]. They share many features of basal-like breast cancer (BLBC) subtype and they are often used synonymously [3]. No targeted therapy exists to treat majority of patients with TNBC because they lack estrogen receptor (ER), progesterone receptor (PR) and HER2/neu [4, 5].
The PI3K/Akt/mTOR pathway is most frequently altered in breast cancers and has been associated with drug resistance [6, 7]. The mechanistic target of rapamycin (mTOR), which acts downstream of PI3K/Akt, is an important target for breast cancer therapy since it is frequently deregulated in breast cancers and plays a critical role in tumorigenesis [8]. mTOR forms complexes with either raptor (mTORC1) or rictor (mTORC2). mTORC1 mediates its function via its downstream targets ribosomal S6 kinase (S6K1 and S6K2) and 4E-binding protein 1 (4E-BP1) [9].
40S ribosomal protein S6 kinase 2 or S6K2 encoded by RPS6KB2 is localized on chromosome 11q13 [10, 11], which constitutes a high-risk subgroup of ER-positive breast cancers [12]. RPS6KB2 gains/amplifications have been associated with poor prognosis and resistance to endocrine therapy [10, 11]. A recent study showed high levels of S6K2 was associated with ER-positive breast cancers [13]. We have shown that S6K2 promotes survival of ER-positive breast cancers to apoptotic stimuli [14].
The PI3K/Akt/mTOR pathway is also frequently deregulated in TNBC [2]. Since chemotherapy continues to be the standard-of-care treatment for TNBC but unacceptable side effects are major problems in the treatment, we examined if targeting S6K2 could enhance sensitivity of TNBCs to chemotherapeutic agents. Our results show that S6K2 is overexpressed in TNBC and depletion of S6K2 sensitized TNBC cells to apoptotic stimuli.
Methods and Materials
Cell Culture
The MCF-10A series developed by Dr. Fred Miller and colleagues [15], was purchased from the Barbara Ann Karmanos Cancer Institute (Detroit, MI). MCF10A, MCF10AT and MCF10AT3G cells were cultured as described previously [16]. MCF-10CA1a and MCF10-CA1d cells were maintained in DMEM-F12 medium supplemented with 5% horse serum. MDA-MB-231 and HCC1395 (obtained from Drs. Adi Gazdar and John Minna, UT Southwestern Medical Center) cells were maintained in RPMI 1640 medium supplemented with 5% fetal bovine serum and 2 mM glutamine. Cells were kept in a humidified incubator at 370C with 95% air and 5% CO2.
Transfection
Cells were transfected with control non-targeting or target-specific siRNAs (GE Dharmacon, Chicago, IL) using Lipofectamine RNAiMax transfection reagent (Invitrogen, Carlsbad, CA) as described before [17]. We overexpressed S6K2 in MDA-MB-231 cells. We purchased pcDNA3 S6K2 E388 D3E (a gift from Dr. John Blenis) from Addgene plasmid#17731; http: //n2t.net/addgene: 17731; RRID: Addgene_17731) [18] and cloned into pLVX-mCherry-C1 lentiviral vector (Clontech). MDA-MB-231 cells were infected with either the empty vector pLVX or vector containing S6K2 construct. The extent of gene knockdown or overexpression was determined by Western blot analysis.
Immunoblot Analysis
Cells were harvested by trypsinization, washed twice with PBS and lysed in buffer containing 20 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 1 mM EGTA, 1 mM EDTA, 1.0% Triton X-100, 0.5% Nonidet-40, 10 mM β-glycerophosphate, protease inhibitor cocktail and phosphatase inhibitor cocktail (Calbiochem/EMD-Millipore, Bedford, MA). Equivalent amounts of total proteins were electrophoresed by SDS-PAGE and transferred electrophoretically to polyvinylidene difluoride membrane (EMD Millipore, Bedford, MA). Immunoblot analyses were performed with 1: 1, 000 dilution of S6K1, P-S6 (Cell Signaling Technology, Danvers, MA) and S6K2 (R&D Systems, Minneapolis, MN) or 1: 5, 000 dilution of PARP (Pharmingen, San Diego, CA) and actin antibodies (Sigma, St. Louis, MO) as described before [19]. 1: 10, 000 dilution of horseradish-peroxidase-conjugated donkey anti-rabbit or goat anti-mouse secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used. The blots were visualized using the enhanced chemiluminescence detection kit (Amersham, Arlington Heights, IL) and the manufacturer’s protocol. The intensities of immunoreactive proteins were quantified using ImageJ software. The blots were probed with actin to control for equal loading.
Immunohistochemistry (IHC)
Formalin-fixed paraffin-embedded tissue microarrays (TMA) obtained from US BioMax, Inc. were deparaffinized with successive washes of xylene and ethanol, and rehydrated. The endogenous peroxidase was blocked by incubation with 0.3% H2O2 and antigen retrieval was performed by heating in citrate buffer (pH 6.0). Immunohistochemistry was performed with anti-S6K2 antibody and immunodetection was performed using a Vecstatin ABC Elite kit and a DAB peroxidase substrate kit from Vector laboratories according to the manufacturer’s protocol.
Caspase activity assay
Cells transfected with control non-targeting or S6K2 siRNA were treated with or without TRAIL. DEVDase activity was determined at 370C using an Ac-DEVD-AFC assay kit (BioVision, Palo Alto, CA, USA) and the manufacturer’s protocol [19]. The fluorescence liberated from DEVD-AFC was measured using a SpectraMax GeminiXS fluorometer and SOFTmax PRO 3.1.1 software (Molecular Devices, Sunnyvale, CA, USA) with an excitation wavelength of 400-nm and emission wavelength of 505 nm.
Annexin V/Propidium Iodide Binding Assay
Cells were treated with or without 0.1 nM TRAIL (R&D Systems, Minneapolis, MN) for 14 h. At the end of the incubation, both detached and attached cells were collected and washed with phosphate-buffered saline. Cells were then stained with annexin V-Alexa 488 conjugate and propidium iodide (Molecular Probes, Eugene, OR) according to the manufacturer’s protocol and analyzed using a flow cytometer (Coulter Epics) [14].
Statistical analysis
Statistical significance was determined by paired Student’s t-test using Microsoft Excel. A p-value < 0.05 was considered statistically significant.
Results
Comparison of S6K2 expression in ER-positive and triple-negative breast cancers
Both S6K1 and S6K2 have been implicated in breast cancer [20, 21]. Therefore, we compared the expression of S6K1 and S6K2 in breast tumor tissues. We analyzed 106 normal and 813 breast tumor tissues from TCGA dataset. The expression of S6K2 but not S6K1 is increased by approximately 2-fold in breast tumor tissues (Fig. 1A), including both ER-positive (n=575) and triple-negative (TN) (n=118) subtypes (Fig 1B). This correlation holds true when tumor tissues are compared with matched normal tissues (Fig. 1C).
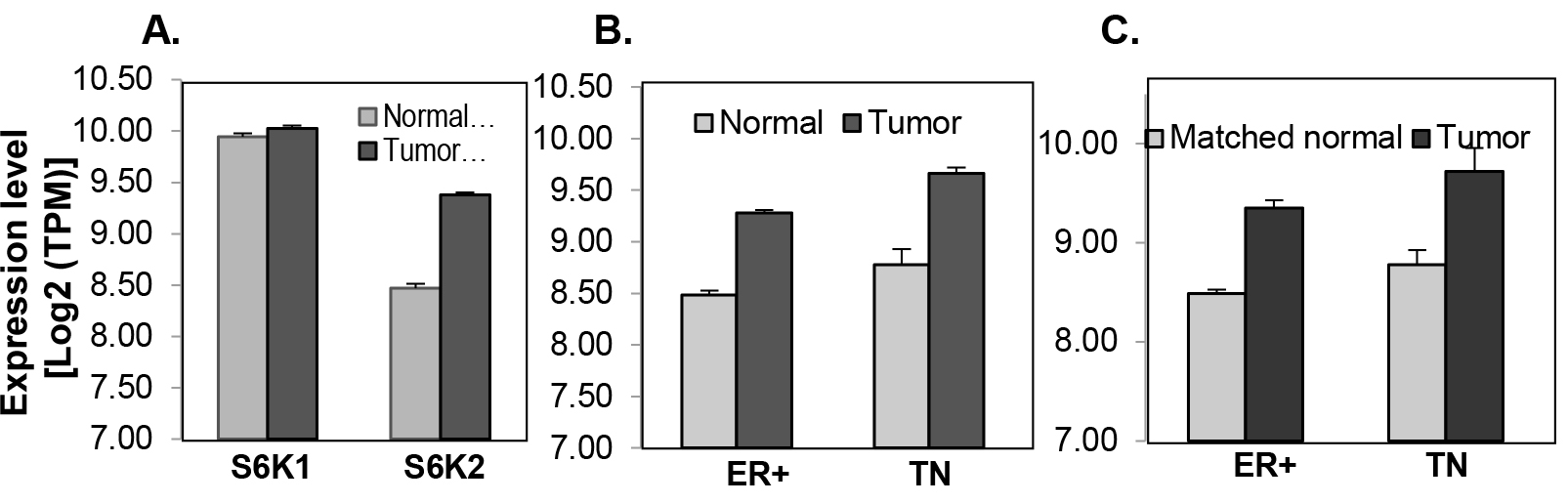
Figure 1. Analysis of S6K1 and S6K2 expression in the TCGA dataset. mRNA expression was measured by RNA-seq in TCGA breast tissue samples. A, S6K1 and S6K2 in all breast tumors (789) and normal (103) samples. B, S6K2 in either ER+ (ER) or triple-negative (TN) subtypes (ER+, N=575, TN, N=118) comparing with those in the normal samples from ER+ (73) or TN (12) patients. C, S6K2 in matched tumor-normal sample pairs of ER+ (73) and TN (12) patients. The error-bar shows the standard deviation of mRNA level. Except for the comparison of S6K1 between normal and tumors samples, all of the comparison for S6K2 levels between tumor and normal samples are significant (p-value < 0.0005), with the fold-change near two (1.74~1.92).
We also analyzed 112 breast tumor specimens and 12 normal tissues in a tissue microarray (TMA) by immunohistochemistry (IHC) (Fig. 2). S6K2 staining is low in normal mammary epithelial cells (Fig. 2A). The staining of S6K2 was intense in both ER-positive (Fig. 2B) and TN (Fig. 2C) breast cancer cells. Thus, analysis of both TCGA and IHC show that S6K2 level is elevated not only in ER-positive breast cancer but also in TNBC.
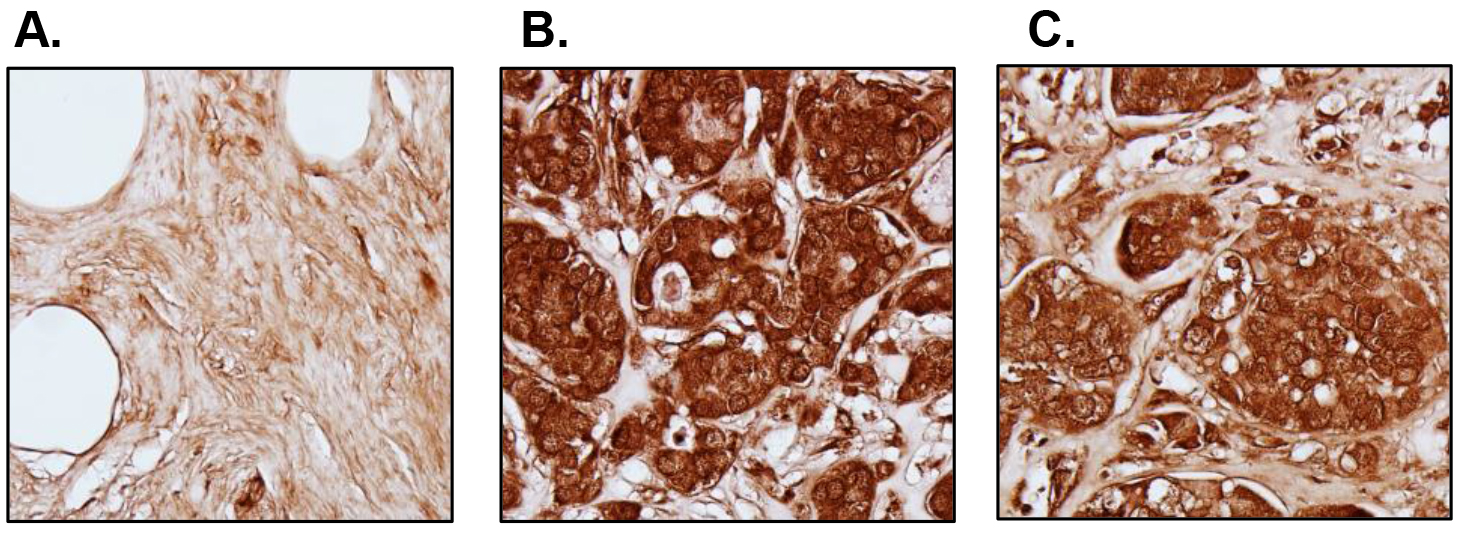
Figure 2. S6K2 expression in normal versus breast tumor tissues. IHC was performed with tissue microarrays. Representative images are shown. A, Cancer adjacent normal breast tissue; B, ER-positive breast tumor (Stage IIb); C, TNBC (Stage IIb). We did not counterstain the slides with hematoxylin since it may obscure S6K2 staining.
We then examined the status of S6K2 in several breast cancer cell lines. We have used progressive basal-like MCF10A series, which include immortalized but non-tumorigenic MCF10A cells as well as highly aggressive metastatic MCF10CA1a and MCF10CA1d variants. These cells do not express hormone receptors. We also included TNBC MDA-MB-231 and ER-positive MCF-7 cells in our comparison. S6K2 but not S6K1 level was increased modestly in MCF10CA1a and MCF10CA1d cells (Fig. 3). In contrast, S6K2 level was low in MDA-MB-231 cells. p70S6K1 gene is amplified in MCF-7 cells and the upper band represents the p85 form of S6K1. Both Akt which acts upstream of mTOR as well as P-S6 which acts downstream of mTORC1/S6K were elevated in MCF10CA1a and MCF10CA1d cells compared to non-tumorigenic MCF10A cells, suggesting that mTORC1 signaling is activated in these cells. In subsequent studies, we depleted S6K2 from MCF10CA1a and MCF10CA1d cells and overexpressed S6K2 in MDA-MB-231 cells.
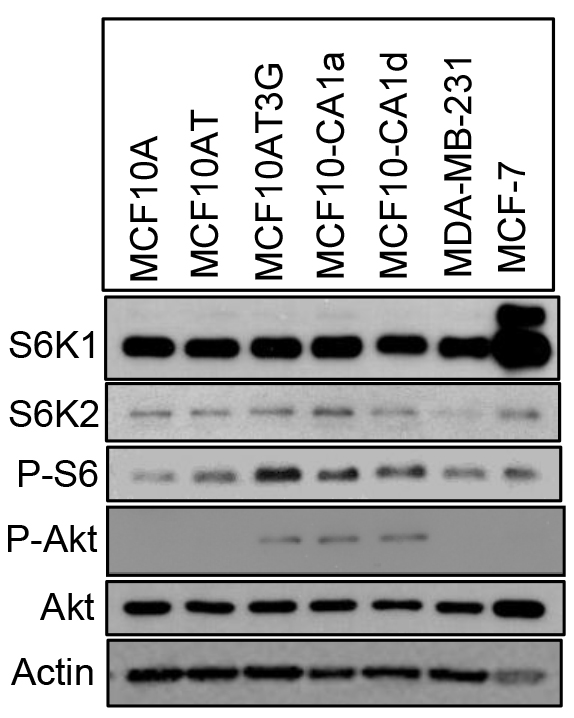
Figure 3. Comparison of Akt and S6K levels in breast cancer cells. Western blot analysis was performed with the indicated antibodies with total cell lysates. The upper band in S6K1 blot represents the p85 form of S6K1.
Effect of S6K2 knockdown on the sensitivity of TNBC cells to apoptotic stimuli
We examined if depletion of S6K2 enhances cell death in TNBC cells by monitoring the cleavage of PARP, a substrate for caspase-3 and -7. Figure 4 shows that while silencing of S6K2 by siRNA alone had little effect on PARP cleavage, it increased the levels of cleaved PARP in response to chemotherapeutic drugs cisplatin and doxorubicin.
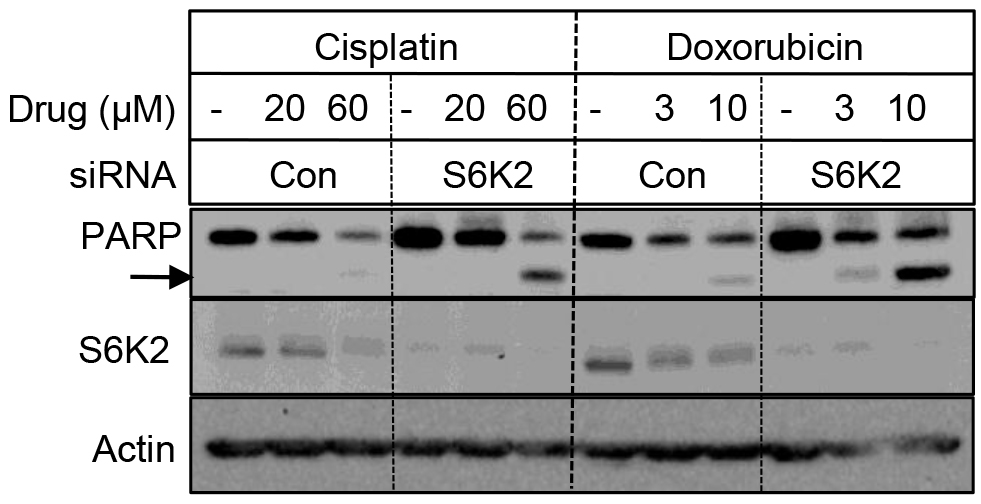
Figure 4. Depletion of S6K2 enhanced cisplatin- and doxorubicin-induced PARP cleavage in MCF10CA1d breast cancer cells. MCF10CA1d cells transfected with control non-targeting or S6K2 siRNAs were treated with or without indicated concentrations of cisplatin or doxorubicin and Western blot analysis was performed with indicated antibodies.
We then examined if depletion of S6K2 increases sensitivity of MCF10CA1a cells to another apoptotic stimulus TRAIL (TNF-related apoptosis-inducing ligand). Knockdown of S6K2 increased TRAIL-induced PARP cleavage as well as the level of cleaved caspase-9 (Fig. 5A), suggesting that S6K2 depletion increased TRAIL-induced apoptosis. To directly determine the effect of S6K2 knockdown on apoptosis, we monitored caspase-3 activity by using fluorogenic DEVD-AFC substrate. Cells transfected with control non-targeting or S6K2 siRNA were treated with TRAIL and the time-course of caspase-3 activation was monitored. As shown in Figure 5B, S6K2 knockdown alone (unfilled triangle) caused a slight increase in caspase-3 activity as compared to control siRNA-transfected cells (unfilled circle). While TRAIL treatment had little effect on caspase-3 activity in control siRNA-transfected cells (filled circle), it dramatically enhanced time-dependent increase in caspase-3 activity in S6K2 siRNA-transfected cells (filled triangle).
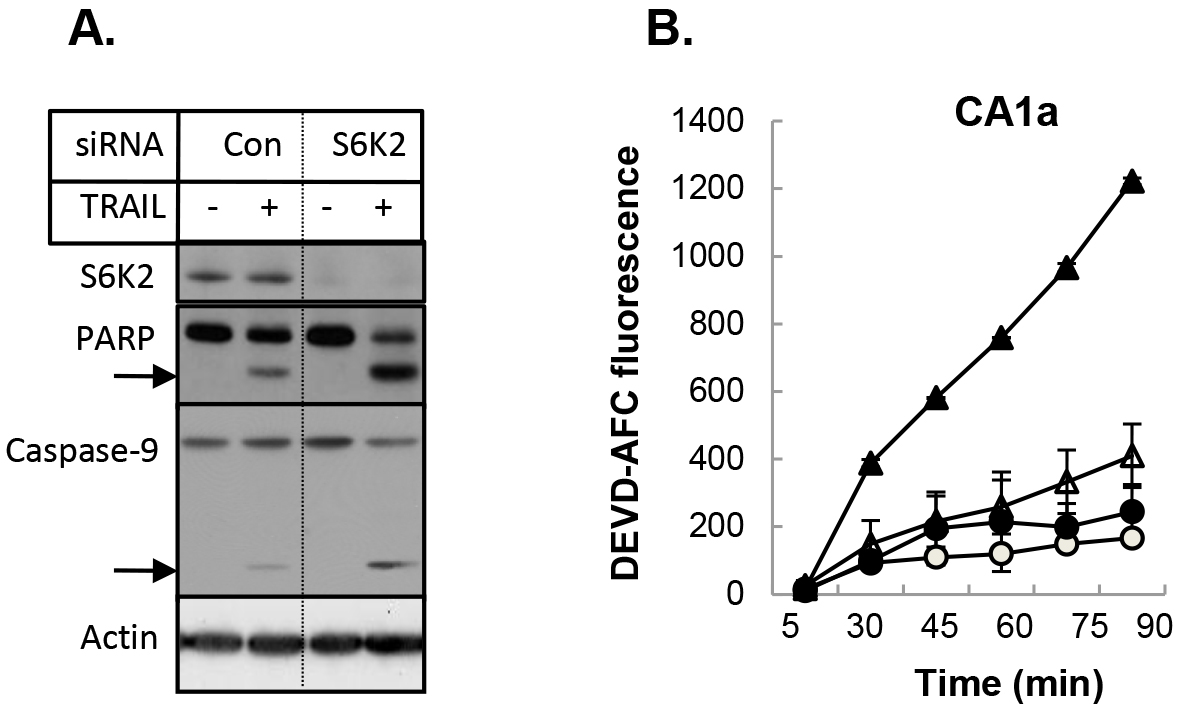
Figure 5. Depletion of S6K2 enhanced TRAIL-induced apoptosis in MCF10CA1a cells. A, MCF10CA1a cells were transfected with control non-targeting or S6K2 siRNA and treated with or without TRAIL. Western blot analysis was performed with indicated antibodies. B, MCF-10CA1a cells transfected with control non-targeting (circle) or S6K2 (triangle) siRNA were treated without (unfilled) or with (filled) TRAIL. Caspase activity assay was performed with DEVD-AFC as the substrate. Fluorescent intensity was calculated for equal amount of protein.
To further examine the effect of S6K2 knockdown on apoptosis, we examined the consequence of S6K2 knockdown on TRAIL-induced apoptosis in HCC1395 cells, which were derived from patients with TNBC. Figure 6 shows that knockdown of S6K2 caused a modest increase in TRAIL-induced PARP cleavage. Similar results were obtained when we monitored apoptosis by Annexin V/PI dye binding assay (Fig. 5B).
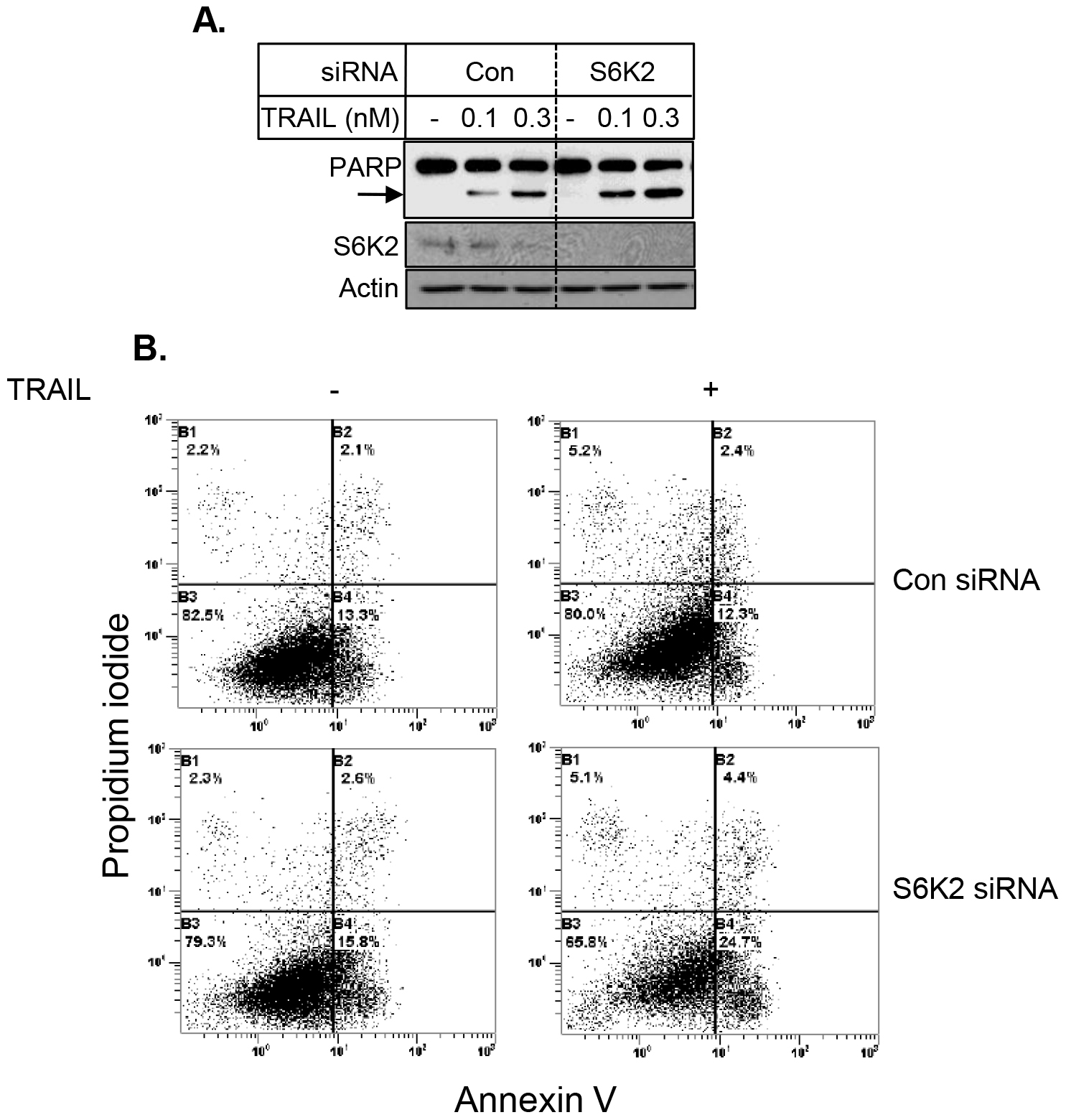
Figure 6. Depletion of S6K2 enhanced TRAIL-induced apoptosis in HCC1395 cells. HCC1395 cells were transfected with control non-targeting or S6K2 siRNA and treated with indicated concentrations of TRAIL. A, Western blot analysis was performed with indicated antibodies. B, Annexin V/PI staining was performed to determine cell death by flow cytometry.
Effect of S6K2 overexpression on the sensitivity of TNBC cells to apoptotic stimuli
To further demonstrate the effect of S6K2 on apoptosis, we overexpressed S6K2 in MDA-MB-231 cells, which express low levels of S6K2. We used an S6K2 construct in which Thr388 was mutated to Glu, and Ser residues at 410, 417 and 423 were mutated to Asp. This phosphomimicking constitutively active form of S6K2 is resistant to mTORC1 inhibitor rapamycin. As shown in Figure 7A, overexpression of S6K2 attenuated TRAIL-induced PARP cleavage. Moreover, while treatment of MDA-MB-231 cells with TRAIL caused a substantial increase in caspase-3 activity, overexpression of S6K2 blunted this increase. These results suggest that S6K2 protects MDA-MB-231 cells against apoptotic stimuli.
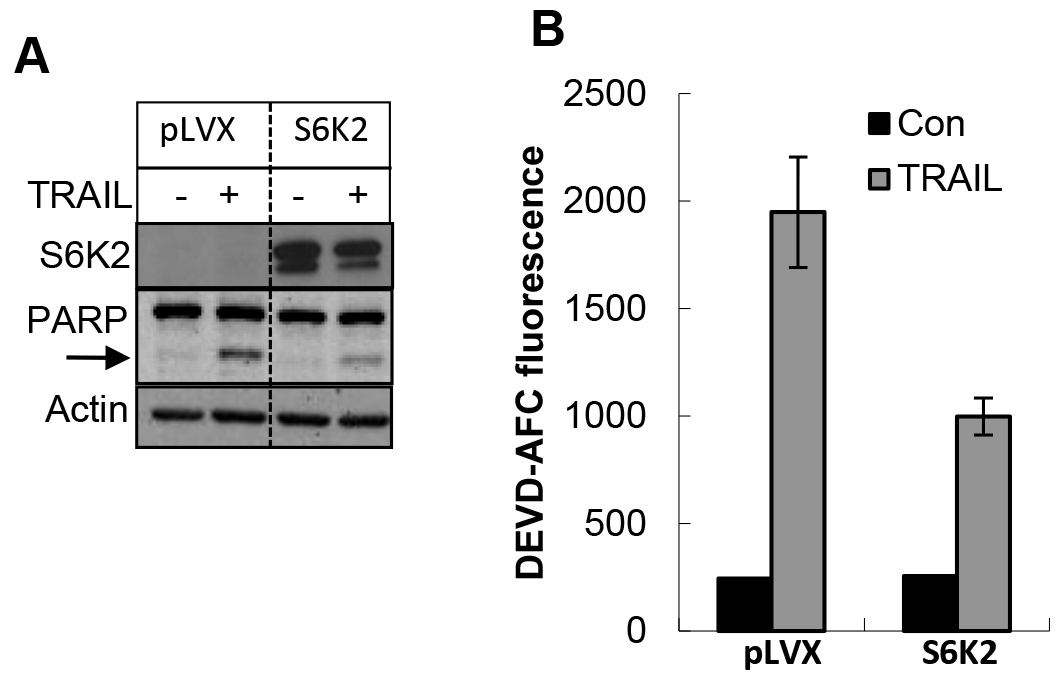
Figure 7. Overexpression of S6K2 attenuated TRAIL-induced apoptosis in MDA-MB-231 cells. MDA-MB-231 breast cancer cells expressing either control vector or rapamycin-resistant (RR) S6K2 were treated with or without TRAIL. A, Western blot analysis was performed with indicated antibodies. B, Caspase activity assay was performed with DEVD-AFC as the substrate.
Discussion
There has been significant advancement in the treatment of breast cancer. The presence of hormone receptors in ER-positive breast cancers suggests a favorable prognosis since the patients can be treated with anti-estrogens or aromatase inhibitors [22]. Similarly, HER2-enriched breast cancers could be treated with herceptin (tratuzumab), a monoclonal antibody against HER2 receptors. TNBCs remain a clinical challenge due to lack of appropriate FDA-approved targeted therapies for most patients with TNBC [3, 4]. Chemotherapy continues to be the standard-of-care treatment for systemic therapy in TNBC patients [23]. However, progression-free survival is short and patients often advance to aggressive metastatic disease [3]. In addition, unacceptable toxicity of cytotoxic chemotherapeutic drugs is a serious problem in the management of the disease.
The PI3K/Akt/mTOR pathway is frequently deregulated in breast cancers and there have been numerous attempts to target this pathway albeit with limited efficacy [24, 25]. While mTORC1 and its downstream target S6K1 have been studied extensively, little attention has been paid to S6K2 [26, 27]. Because of the high degree of homology between S6K1 and S6K2, it was generally believed that the function of these two homologs is similar [26, 27]. Emerging studies identified S6K2 as a novel candidate oncogene and overexpression of S6K2 was associated with poor prognosis and resistance to endocrine therapy [10, 11]. We have shown that S6K1 and S6K2 have opposite effect on the survival of ER-positive breast cancers [14]. Knockdown of S6K2 but not S6K1 enhanced cell death in response to apoptotic stimuli in ER-positive breast cancer cells [17].
The results of our present study show that S6K2 also plays an important role in promoting the survival of TNBC. Based on the analysis of the TCGA dataset, the expression of S6K2 but not S6K1 is increased in both ER-positive and TN breast cancers. Consistent with the TCGA data, immunohistochemical staining of patient samples also revealed that S6K2 protein expression was higher in both ER-positive and TN subtypes. Moreover, in the progressive MCF10A breast cancer model, S6K2 levels appear to be higher in aggressive cell lines compared to non-malignant cells. However, S6K2 level was low in MDA-MB-231 cells, which may rely on other oncogenic signaling pathways for survival.
We found that depletion of S6K2 alone had little effect on cell death but S6K2 knockdown enhanced cell death in MCF10ACA1a, MCF10CA1d and HCC1395 cells that lack ER, PR and HER2/neu. We have used chemotherapeutic drugs, such as doxorubicin and cisplatin that act via the mitochondrial pathway as well as TRAIL, which acts via the receptor-initiated pathway. S6K2 knockdown enhanced cell death by apoptosis as evident by the cleavage of PARP, caspase-3 activity and annexin V/PI staining. Furthermore, overexpression of S6K2 in MDA-MB-231 cells attenuated TRAIL-induced apoptosis reinforcing our notion that S6K2 promotes survival in TNBC cells.
The PI3K/Akt and Ras/MAPK pathways are often deregulated in breast cancers [22, 30]. We have previously shown that S6K2 promotes survival of ER-positive breast cancer MCF-7 cells partly via activation of the Akt signaling pathway [14]. It remains to be seen if S6K2 promotes survival of TNBC cells via Akt-dependent or -independent pathway. P-Akt could be detected in malignant MCF10ACA1a and MCF10CA1d cells but not in non-malignant MCF10A cells. During the generation of MCF-10CA1a cells, MCF10A cells were transfected with T24 Ha-Ras [15]. Therefore, ERK signaling pathway is also activated in these cells. Thus, the observation that S6K2 KD induced cell death in MCF10CA1a and MCF10CA1d cells with activated Akt/mTOR and Ras/Raf/MAPK pathway [16] is significant.
Given that chemotherapy continues to be the major treatment option for TNBC patients and S6K2 knockout mice are viable and exhibit normal development and homeostasis [26, 28–30], intervention with S6K2 in combination with other chemo- or biological therapeutic agents is expected to provide a novel non-toxic therapy for TNBCs.
Author Contributions: AB and SS designed and performed experiments, ZX analyzed TCGA dataset, AB wrote the manuscript and SS edited the manuscript.
Abbreviations:
BC, Breast cancer;
BLBC, Basal-like breast cancer;
4E-BP1, Eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1;
ERK, Extracellular signal-regulated kinase;
ER, Estrogen receptor;
HER2, Human epidermal growth factor receptor 2;
IHC, Immunohistochemistry;
mTORC1, Mechanistic target of rapamycin complex 1;
PARP, Poly (ADP-ribose) polymerase;
PI, Propidium iodide;
PI3K, Phosphatidylinositol-3-kinase;
PR Progesterone receptor;
p70 S6 kinase 1 (S6K1);
S6K2, 40S ribosomal protein S6 kinase 2;
TCGA, The cancer genome atlas;
TNBC, Triple-negative breast cancer;
TMA, Tissue microarray;
TNF, Tumor necrosis factor-α;
TRAIL, TNF-related apoptosis-inducing ligand;
References
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000; 406: 747–52.
- Costa RLB, Han HS, Gradishar WJ. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: a review. Breast cancer research and treatment. 2018; 169: 397–406.
- Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nature reviews Clinical oncology. 2010; 7: 683–92.
- Bayraktar S, Gluck S. Molecularly targeted therapies for metastatic triple-negative breast cancer. Breast cancer research and treatment. 2013; 138: 21–35.
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. The New England journal of medicine. 2010; 363: 1938–48.
- Bahrami A, Khazaei M, Shahidsales S, Hassanian SM, Hasanzadeh M, Maftouh M, et al. The Therapeutic Potential of PI3K/Akt/mTOR Inhibitors in Breast Cancer: Rational and Progress. Journal of cellular biochemistry. 2018; 119: 213–22.
- Guerrero-Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer metastasis reviews. 2016; 35: 515–24.
- Mita MM, Mita A, Rowinsky EK. Mammalian target of rapamycin: a new molecular target for breast cancer. Clinical breast cancer. 2003; 4: 126–37.
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nature Reviews Cancer. 2006; 6: 729–34.
- Karlsson E, Perez-Tenorio G, Amin R, Bostner J, Skoog L, Fornander T, et al. The mTOR effectors 4EBP1 and S6K2 are frequently coexpressed, and associated with a poor prognosis and endocrine resistance in breast cancer: a retrospective study including patients from the randomised Stockholm tamoxifen trials. Breast cancer research : BCR. 2013; 15: R96.
- Karlsson E, Waltersson MA, Bostner J, Perez-Tenorio G, Olsson B, Hallbeck AL, et al. High-resolution genomic analysis of the 11q13 amplicon in breast cancers identifies synergy with 8p12 amplification, involving the mTOR targets S6K2 and 4EBP1. Genes Chromosomes Cancer. 2011; 50: 775–87.
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2, 000 breast tumours reveals novel subgroups. Nature. 2012; 486: 346–52.
- Bostner J, Karlsson E, Eding CB, Perez-Tenorio G, Franzen H, Konstantinell A, et al. S6 kinase signaling: tamoxifen response and prognostic indication in two breast cancer cohorts. Endocrine-related cancer. 2015; 22: 331–43.
- Sridharan S, Basu A. S6 kinase 2 promotes breast cancer cell survival via Akt. Cancer research. 2011; 71: 2590–9.
- Santner SJ, Dawson PJ, Tait L, Soule HD, Eliason J, Mohamed AN, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast cancer research and treatment. 2001; 65: 101–10.
- Kim SH, Miller FR, Tait L, Zheng J, Novak RF. Proteomic and phosphoproteomic alterations in benign, premalignant and tumor human breast epithelial cells and xenograft lesions: biomarkers of progression. International journal of cancer. 2009; 124: 2813–28.
- Basu A, Sridharan S. Regulation of anti-apoptotic Bcl-2 family protein Mcl-1 by S6 kinase 2. PLoS One. 2017; 12: e0173854.
- Lee-Fruman KK, Kuo CJ, Lippincott J, Terada N, Blenis J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene. 1999; 18: 5108–14.
- Basu A, Akkaraju GR. Regulation of caspase activation and cis-diamminedichloroplatinum(II)-induced cell death by protein kinase C. Biochemistry. 1999; 38: 4245–51.
- Barlund M, Monni, O., Kononen, J., Cornelison, R., Torhorst, J., Sauter, G., Kallioniemi, O.P., and Kollioniemi, A. Multiple Genes at 17q23 Undergo Amplification and Overexpression in Breast Cancer. Cancer Res. 2000; 60: 5340–4.
- Perez-Tenorio G, Karlsson E, Waltersson MA, Olsson B, Holmlund B, Nordenskjold B, et al. Clinical potential of the mTOR targets S6K1 and S6K2 in breast cancer. Breast cancer research and treatment. 2010.
- Yamamoto-Ibusuki M, Arnedos M, Andre F. Targeted therapies for ER+/HER2- metastatic breast cancer. BMC Med. 2015; 13: 137.
- Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nature reviews Clinical oncology. 2016; 13: 674–90.
- Dey N, De P, Leyland-Jones B. PI3K-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol Ther. 2017; 175: 91–106.
- Sivendran S, Agarwal N, Gartrell B, Ying J, Boucher KM, Choueiri TK, et al. Metabolic complications with the use of mTOR inhibitors for cancer therapy. Cancer Treat Rev. 2014; 40: 190–6.
- Pardo OE, Seckl MJ. S6K2: The Neglected S6 Kinase Family Member. Front Oncol. 2013; 3: 191.
- Tavares MR, Pavan IC, Amaral CL, Meneguello L, Luchessi AD, Simabuco FM. The S6K protein family in health and disease. Life Sci. 2015; 131: 1–10.
- Fenton TR, Gout IT. Functions and regulation of the 70kDa ribosomal S6 kinases. The international journal of biochemistry & cell biology. 2011; 43: 47–59.
- Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, et al. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5’-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Molecular and cellular biology. 2004; 24: 3112–24.
- Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. The EMBO journal. 1998; 17: 6649–59.