Abstract
In silico methods can help in identifying drug targets via bioinformatics tools. In our previous studies, a 2.34 kDa short linear peptide analogue to myxoma virus M-T5 protein was computationally designed using the resonant recognition model (RRM). Quantitative and qualitative cell cytotoxicity assays showed that the peptide analogue can induce a dose and time dependent tumoricidal effect on skin cancer cells but not on normal cells. It has been reported that the viral protein (M-T5) binds with p-Akt to regulate Akt signaling in some human cancer types. Hence in this study we investigated the effects induced by the peptide analogue RRM- MV on the Akt pathway in skin cancer and normal cells. Akt expression levels in cells treated with RRM-MV, in the presence or absence of PI3K/Akt inhibitor, were detected in mammalian skin cancer and normal cells using immunoblotting. The results revealed that different endogenous levels of p-Akt were expressed in the human melanoma and carcinoma cells following treatment with RRM-MV. Yet, it did not appear to affect the p-Akt expression in the mouse melanoma cells. Furthermore, RRM-MV treatment did not seem to affect the level of total Akt in any type of cancer cells under the same experimental conditions. Hence, the bioactive peptide RRM-MV appears to be targeting the Akt pathway in some types of skin cancer cells. Elucidating the mechanism of RRM-MV effects on other cellular apoptosis pathways requires further investigation.
Keywords
Akt, RRM-MV, Cell death pathway, Carcinoma, Melanoma, Myxoma virus.
Background
Malignant melanomas can be resistant to conventional therapies such as chemotherapy, immunotherapy, radiation therapy and/or surgery, and patient’s chances of survival often depend on early diagnosis and treatment [1]. The discovery and development of novel cancer therapeutic agents is an important research field as cancer is an international health problem with the current treatments achieving only limited success. Oncolytic virotherapy is a novel therapy that can be applied solely or in conjunction with the conventional therapies. In particular, some viruses have the ability to target and destroy malignant cancer cells without harming the normal cells [2- 5]. Myxoma virus (MYXV) is a new poxvirus oncolytic candidate that has been added to the list of oncolytic viruses used in virotherapy. MYXV is a member of Poxviridae family and the Chordopoxvirinae subfamily [6-8]. It is a rabbit-specific pathogen that causes myxomatosis in European rabbits (Oryctolagus cuniculus), which is considered as a fatal disease [8]. However, MYXV is non-pathogenic in humans and all other vertebrate species [6, 9-13].
Previously, MYXV was shown to be able to selectively kill human cancer cells [14-17]. Intense in vitro investigations have indicated that replication of MYXV in human cancer cells is associated with the hyperactivation of the serine/threonine kinase Akt in cancer cells [17-27]. In addition, human cancer cells have been grouped into three types depending on the ability of being productively infected with MYXV, and the capability of MYXV to activate Akt through the action of a M-T5 viral protein [26, 28]. M-T5 is an ankyrin-repeat host range protein that is shown to be required for virus replication in rabbit lymphocytes, and is critical for virus replication in the majority of human tumor cells [29]. M-T5 interacts with two human cellular proteins. The first cellular protein is Cullin-1, an E3 ubiquitin ligase that is involved in the progression through the cell cycle. The interaction of M-T5 with Cullin-1 inhibits MYXV- infected cells from cell cycle arrest and stress-induced cell death [30]. The second cellular protein is Akt-1/protein kinase B (PKB) that directly interacts with M-T5 [26, 31]. Akt is a serine/threonine kinase essential for normal cell processes such as programmed cell death, proliferation, cell cycle progression, angiogenesis and metabolism [22]. It is activated via the phosphorylation of its central component at ser-473/474 and thr-308/309 [32]. Akt dysregulation is commonly detected in an extensive spectrum of human cancers [23, 33-39]. It was found that many human cancer cells exhibit a high endogenous activation of Akt in vitro [18, 33, 34, 40]. Akt is a central regulator of cellular signaling; hence, it is not surprising that many viruses have developed new strategies to control the activation of Akt [41].
Therapeutic peptides have been emerging as a novel class of drugs for cancer therapy and are being intensively studied for their role in developing novel cancer therapeutics [2, 3]. This is due to the fact that peptides have a high potential to penetrate cellular membranes, interfere with enzymatic functions and protein-protein interactions within cells [4-7]. Anticancer therapeutic peptides are able to kill cancer cells due to their strong tumoricidal activity and low toxicity towards normal cells [42-47]. Peptide therapy has many promising advantages over small molecule drugs. Peptides can be designed to target any protein of interest using ‘rational’ computational methods. Due to structures and interactive partners of many oncogenic proteins being identified, peptides can be designed to inhibit these interactions by using a sequence from the interaction domain. Another advantage is that peptides are easily produced using chemical synthesis or molecular biology techniques. Unlike natural proteins, peptides are easily synthesized in bulk at relatively low cost, using automated systems. The costs of producing therapeutic proteins through genetically modified organisms and cell cultures are relatively high.
The Resonant Recognition Model (RRM) [42] is the physico-mathematical approach designed for functional and structural analysis of proteins and DNA. The RRM approach interprets a protein’s sequence linear information using signal analysis methods. In the RRM, the protein primary structure is represented as a numerical series by assigning to each amino acid in the sequence a physical parameter value relevant to the protein’s biological activity. The RRM concept is based on the finding that there is a significant correlation between spectra of the numerical presentation of amino acids and their biological activity. It has been found through an extensive research that proteins with the same biological function have a common frequency in their numerical spectra. This frequency was found then to be a characteristic feature for protein biological function or interaction [43-48]. This protein characteristic frequency can be identified from analysis of the power spectra of the selected protein sequences. In addition, from the analysis of their phase spectra, we can identify the corresponding phase for a particular frequency. Once the characteristic frequency for a particular protein function/or interaction is determined, it is possible then to predict the amino acids, the so called “hot spots” in the sequence that predominantly contribute to this frequency and consequently to the observed function. On the basis of determined RRM characteristic frequencies and phases for a particular group of protein sequences, we can design amino acid sequences (short peptides) having those specific characteristics related to a protein’s biological function. It is expected that the designed peptide will exhibit the desired biological activity [42, 49-50].
In the previous research studies, the RRM approach was used to structure–function analysis of different protein examples [42, 50], aiming at the design of peptide analogues having the same functional activity as their parent proteins. Myxoma virus (MXYV), interleukin12 (IL12) and the tumour necrosis factor (TNF-α) can significantly affect tumour progression. Based on these properties, three short bioactive peptide analogues: RRM-MV RRM-IL and RRM-TNF [51] were designed using the RRM approach [50]. These synthetic peptides were de novo designed to mimic activities of the selected therapeutic proteins. RRM–designed peptides present potential therapeutic candidates for cancer treatment whose efficacies were evaluated in our previous studies [50]. In particular, we examined the biological effects of these peptides on B16F0 cell mouse skin cancer cells and normal murine cells in vitro, and found that RRM-MV, RRM-IL and RRM-TNF treatment induced toxic effects on the mouse melanoma cells in a dose- and time-dependent manner but no cytotoxic effects were detected in normal cells [43-48, 50]. However, our initial data did not show a significant effect on the total Akt levels following treatment of the mouse melanoma cells (B16F) with the peptide analogue RRM-MV[43]. Thus, it is not known if the cytotoxic effects on cancer cells caused by the MT5-peptide analogue (RRM-MV) were generated by targeting the same cell death pathways in skin cancer cells. Therefore, in this study we tested the effect of the RRM-MV and non- bioactive (control) peptide RRM-C on the p-Akt pathway in human skin cancer and normal human skin cells.
Materials and Methods
RRM peptide sequences and synthesis
The RRM-MV is a 18 amino acid short linear peptide (MDDRWPLEYTDDTYEIPW) analogue for myxoma virus M-T5 protein which was specifically designed to exhibit anti-cancer activity [43, 44]. The RRM-C is a 22 amino acid short linear peptide (CVLQDCVLQDCVIQDCVLQDCV) designed as a control peptide that is lacking the anti- cancer activity. It was used as a negative control peptide along with the bioactive RRM-MV in the experimental evaluation presented below. The detailed description of the de novo design procedure for RRM-MV and RRM-C peptides was presented in our previous study [43]. The two peptides were synthesized with >95% purity (AUSPEP, Melbourne, Australia). RRM- MV and RRM-C were freshly prepared at different concentrations in the relevant cell culture medium immediately prior to use.
Mammalian cell culture growth conditions and treatment with the RRM peptides
Human malignant melanoma (MM96L), human squamous cells carcinoma (COLO-16) and normal human dermal fibroblast (HDF) cell lines were obtained from Dr. Terrence Piva, RMIT University, Australia. Mouse melanoma cell line (B16F0) was donated by Dr. Glen Boyle, QIMR, Australia. The MM96L, COLO-16 and B16-F0 cell lines were cultured in Roswell Park Memorial Institute medium (RPMI) 1640 (GIBCO, Australia) supplemented with 10% heat- inactivated fetal bovine serum (FBS) (Bovogen serum, Biologicals, Australia). The HDF cell line was propagated in medium 106 supplemented with 2% Low Serum Growth Supplement (LSGS) (GIBCO, Australia). The cell lines were confirmed to be mycoplasma free using a Mycofluor Detection Kit (Invitrogen, Oceania). All cell cultures were grown and maintained as monolayers in 75 m2 tissue culture flasks (Greiner Bio-One, Germany) at 37°C in a humidified atmosphere and 5% CO2 in air. The medium was changed every 3 – 4 days.
At 85–95% confluence, the cells were harvested by trypsinisation with 0.05% trypsin-EDTA (GIBCO, Australia) and centrifuged. The cell suspensions were seeded at a density of 7×105 cell/mL in a six-well plate in complete growth medium with 10% FBS and incubated overnight before treatment to allow for their adherence. The peptide treatment conditions of cell lines were performed as described below:
Incubation conditions to assess Akt expression levels: To assess the influence of treatment on Akt activity MM96L, COLO-16 and HDF cell lines were incubated with 400 ng/mL RRM-M or RRM-C for 3 h in complete growth medium with 10% FBS. The mouse melanoma B16F0 cell line was incubated with 800 ng/mL RRM-MV or RRM-C for 3 h to achieve cytotoxicity levels similar to those produced in the human skin cancer cell lines within the same period of incubation as this cell line was more resistant to RRM-MV treatment [43, 44].
Treatment with the PI3 Kinase inhibitor
The influence of the RRM-MV treatment on Akt activity, after blocking the Akt pathway, was assessed to confirm if treatment with inhibitors can affect the phosphorylation of Akt at Ser-473 or Thr-308 in the presence of RRM-MV. The effect of RRM-MV and RRM-C on Akt activity was assessed in melanoma, carcinoma and normal cell lines in the presence of the PI3 Kinase inhibitor, LY294002 which blocks the Akt pathway. The cell lines were incubated with 50-100 µM of LY294002 (PI3 Kinase inhibitor) for 1 h-2 h before the RRM-MV or the RRM-C peptides were added and then incubated for 3 h, as explained earlier. This experiment was performed in the presence of a complete growth medium with 10% FBS or in serum deprivation conditions to eliminate the effect of serum on the experimental conditions.
Furthermore, Akt expression levels in MM96L and COLO-16 cell lines, following treatment with 400 ng/mL RRM-MV, were assessed in serum starvation conditions for 15 min, 30 min and 60 min.
Protein extraction for immunoblotting
Following incubation with the RRM-designed peptides (RRM-MV or RRM-C) and/or the PI3 kinase inhibitor, the cells were rinsed twice with ice-cold phosphate buffered saline (PBS) and were then lysed by PhosphoSafe Extraction Buffer (Novagen, USA) mixed with Protease Inhibitor Cocktails (Sigma-Aldrich, USA). The cells were incubated for 10 min at room temperature and were scraped and centrifuged at 16,000x g for 5 min at 4°C. The supernatants were collected and the whole-cell lysates and protein concentration of each sample was determined by Bradford method [51] using bovine serum albumin (BSA) as a standard.
Antibodies and reagents
Total Akt protein, threonine-phosphorylated Akt and serine-phosphorylated Akt were detected using rabbit polyclonal Akt (pan) (C67E7) antibody, rabbit monoclonal phospho-Akt (Thr-308) (C31E5E) antibody and rabbit monoclonal phospho-Akt (Ser-473) (D9E) XP antibody respectively. The β-actin antibody was used as a loading control. All primary antibodies and the PI3K-AKT kinase inhibitor (LY294002) were obtained from Cell Signaling Technology (Beverly MA, USA). The alkaline phosphatase-conjugated goat anti-rabbit polyclonal secondary antibody was purchased from Sapphire Bioscience, Australia.
Western blot analysis
Whole cell lysates were mixed with 5X SDS-PAGE sample buffer (3: 1), consisting of 60 mM Tris, pH 6.8, 25% glycerol, 2% sodium dodecyl sulfate (SDS), 14.4 mM 2-mercaptoethanol, and 0.1% bromophenol blue, and were boiled for 5 min. Equal amounts of protein samples (30µg per lane) were subjected to 12.5% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) using Mini-PROTEAN Tetra Cell electrophoresis system (Bio-Rad, USA). iBlot Dry Blotting System (Invitrogen, USA) was used to transfer the protein from the SDS PAGE gel to the nitrocellulose membrane. Western blot analysis was carried out as described previously [43] with some modifications. In brief, the membranes were blocked with 5% non-fat dry milk in PBS-T (0.1% Tween 20 in PBS) overnight at 4°C. The membranes were then washed four times for 10 min with PBS-T, followed by incubation of the membranes with the primary antibody in (1X Tris Buffered Saline (TBS), 0.1% Tween-20 with 5% BSA) at 1: 3000 dilutions for 3 h, and washed four times for 15 min. The membranes were then incubated with the secondary antibody conjugated with Horseradish Peroxidase in (1X TBS, 0.1% Tween-20 with 0.1% non-fat dry milk) at 1: 1000 dilutions for 1h at room temperature. After washing with PBS-T four times for 15 min, detection of protein was performed using 5-Bromo-4-chloro-3-indolyl phosphate, toluidine salt / Nitro blue tetrazolium chloride (BCIP/NBT) substrate solution (Amresco, USA) mixed with detection buffer (100mM Tris, 100Mm NaCl, pH 9.5) (v/v). The membranes were incubated with the substrate until full color development was detected. The reaction was stopped by washing in distilled water for 5 min prior to being dried. The membranes were scanned to quantitate the signal using a flatbed optical scanner (Cannon, USA). Densitometry of protein spot signals were detected and quantified with the Quantity One Analysis 4.3.0 software. Each experiment was repeated at least three times and the mean of the density of the bands (densitometry signals) was used to plot the graphs in the figures.
Results
The influence of RRM-MV treatment on Akt activity
To determine whether RRM-MV or RRM-C treatments modulate Akt activity in skin cancer cells and normal skin cells, the total Akt protein levels and the endogenous phosphorylated Akt levels at Thr-308 and Ser-473 were measured using Western blots. Results are presented and described in details below.
In presence or absence of serum in the growth medium: The levels of p-Akt and total Akt proteins were detected in 10% serum (FBS) or in serum starvation in MM96L human melanoma cells (Fig 1 A & B), COLO-16 human carcinoma cells (Figure. 2, A & B) and in B16F0 mouse melanoma cells (Figure3, A & B) following RRM-MV or RRM-C treatment. In MM96L cell line, the 60 kDa p-Akt bands’ intensity for Ser-473 antibodies was slightly stronger than for Thr-308 (Figure 1A) under 10% FBS growth conditions. Similarly, p-Akt band intensities were slightly stronger for Ser-473 in COLO-16 cell line under 10% FBS (Figure 2A). Although the expression levels of p-Akt (Ser-473) were slightly increased, following RRM-MV treatment in melanoma and carcinoma cell lines, the results also suggest that the expression levels of total Akt and p-Akt at Ser-473 and Thr-308 phosphorylation locations in serum starved or in complete growth medium were unaffected in the MM96L melanoma cells or the COLO 16 carcinoma cells. Interestingly, RRM-C treatment did not induce any noticeable effect on the p-Akt and total Akt protein expression levels as those remained unchanged in the MM96L cell line (Figures 1A and B, lane 2), COLO-16 cell line (Figures 2A and B, lane 3), and B16F0 cell line (Figure 3, lane 3); thus strongly indicating that the RRM-C treatment has no effect on the Akt expression in these cancer cell lines. We have previously reported that RRM-C control peptide did not cause any cytotoxic effect as opposed to the cytotoxicity induced by RRM-MV treatment on the tested skin cancer cell lines [43; 44]. The Akt expression results could be a reflection of this effect on this cell death pathway.
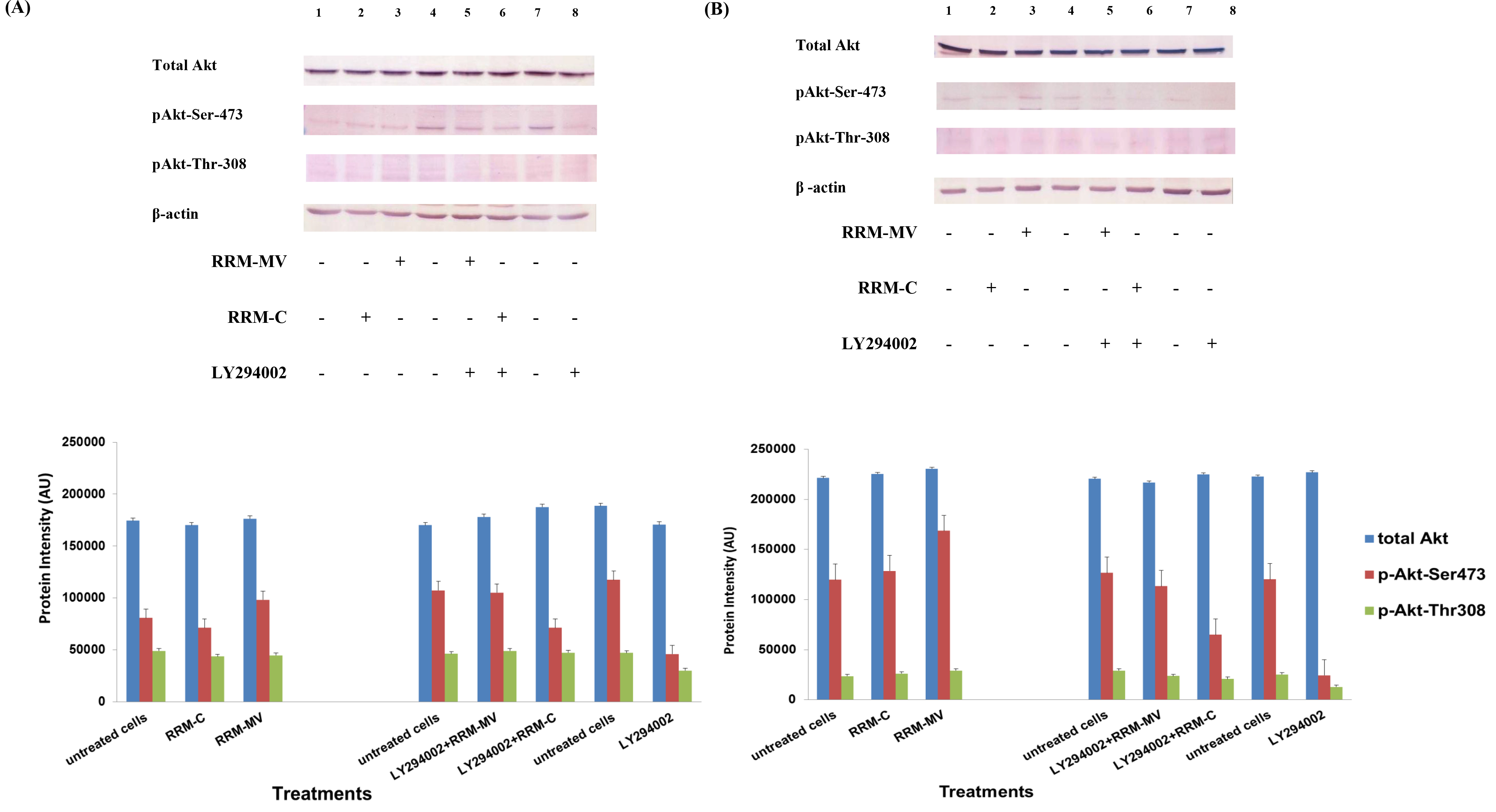
Figure 1. The effects of RRM designed peptides on cellular expression of Akt in melanoma cells. The MM96L cell line was plated at 5 × 107 cells/mL and was treated in presence of serum (A) or in absence of serum (B). Cell lysates (20µg) were prepared and immunoblotted using p- Akt (Ser-473 and Thr-308) and total Akt antibodies. β-actin was used as a loading control. The Western blots results show that treatment with RRM-MV, but not RRM-C, slightly induces the levels of p-Akt in both conditions. Inhibition of Akt activity and then treatment with RRM-MV also affected the expression of p-Akt protein in tested cells. Densitometry of protein spot signals were detected and quantified with the Quantity One Analysis 4.3.0 software. These figures are representative of three separate Western blot experiments.
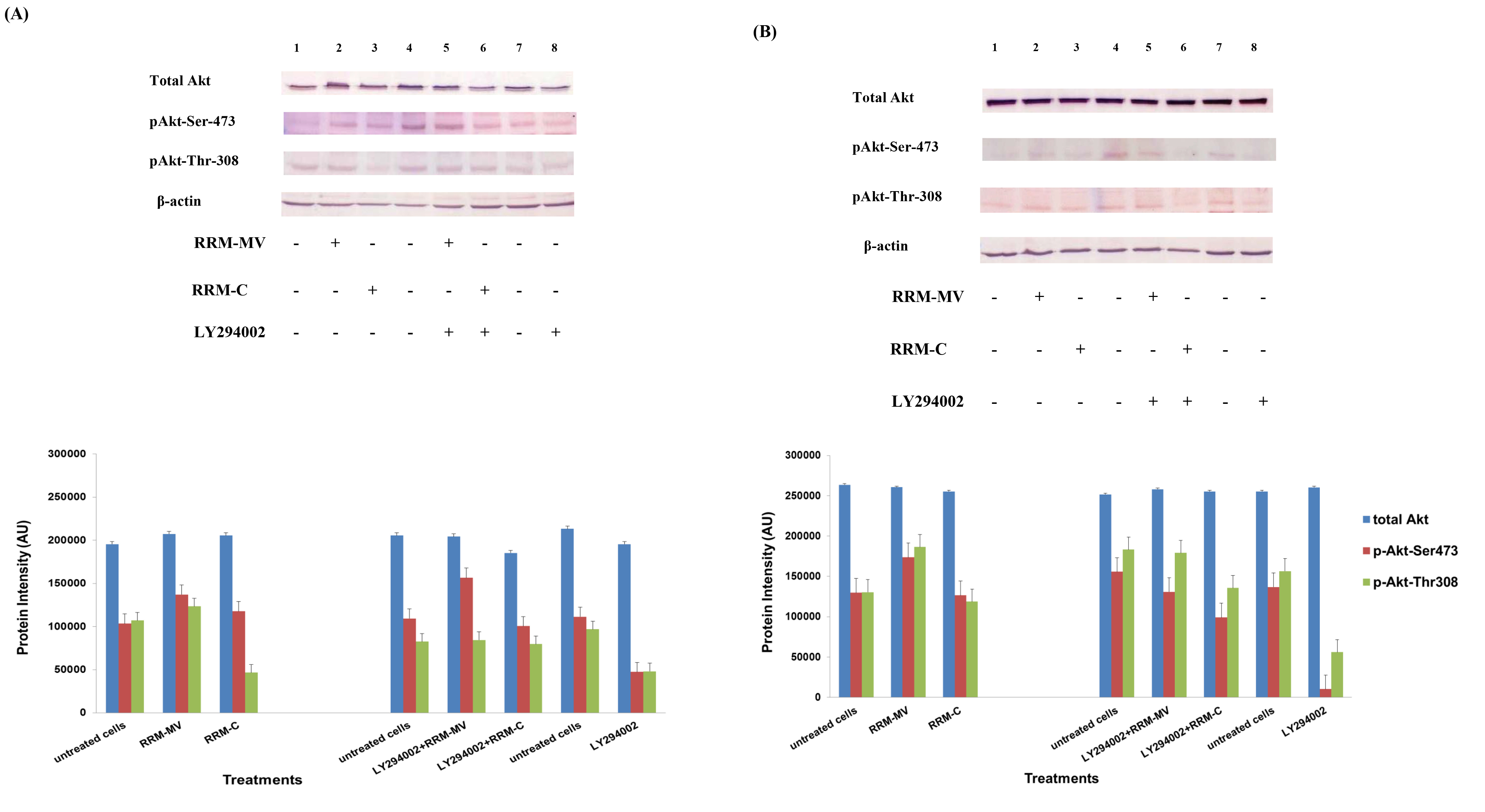
Figure 2. Cellular expression of p-Akt and total Akt in carcinoma cells after treatment with the RRM designed peptides. The COLO-16 cell line was plated at 5 × 107 cells/mL and was treated in growth medium with serum (A) or in serum starvation conditions (B). Western blot analysis was used to assess the expression of Akt in treated cells using p-Akt (Ser-473 and Thr-308) and total Akt antibodies. β-actin was used as a loading control. The Western blot results show that the expressions of p-Akt in carcinoma cells slightly increased after RRM-MV treatment. Inhibition of Akt activity and then treatment with RRM-MV also affected the expression of p-Akt protein in tested cells. Densitometry of protein spot signals were detected and quantified with the Quantity One Analysis 4.3.0 software. The experiments were repeated at least three times.
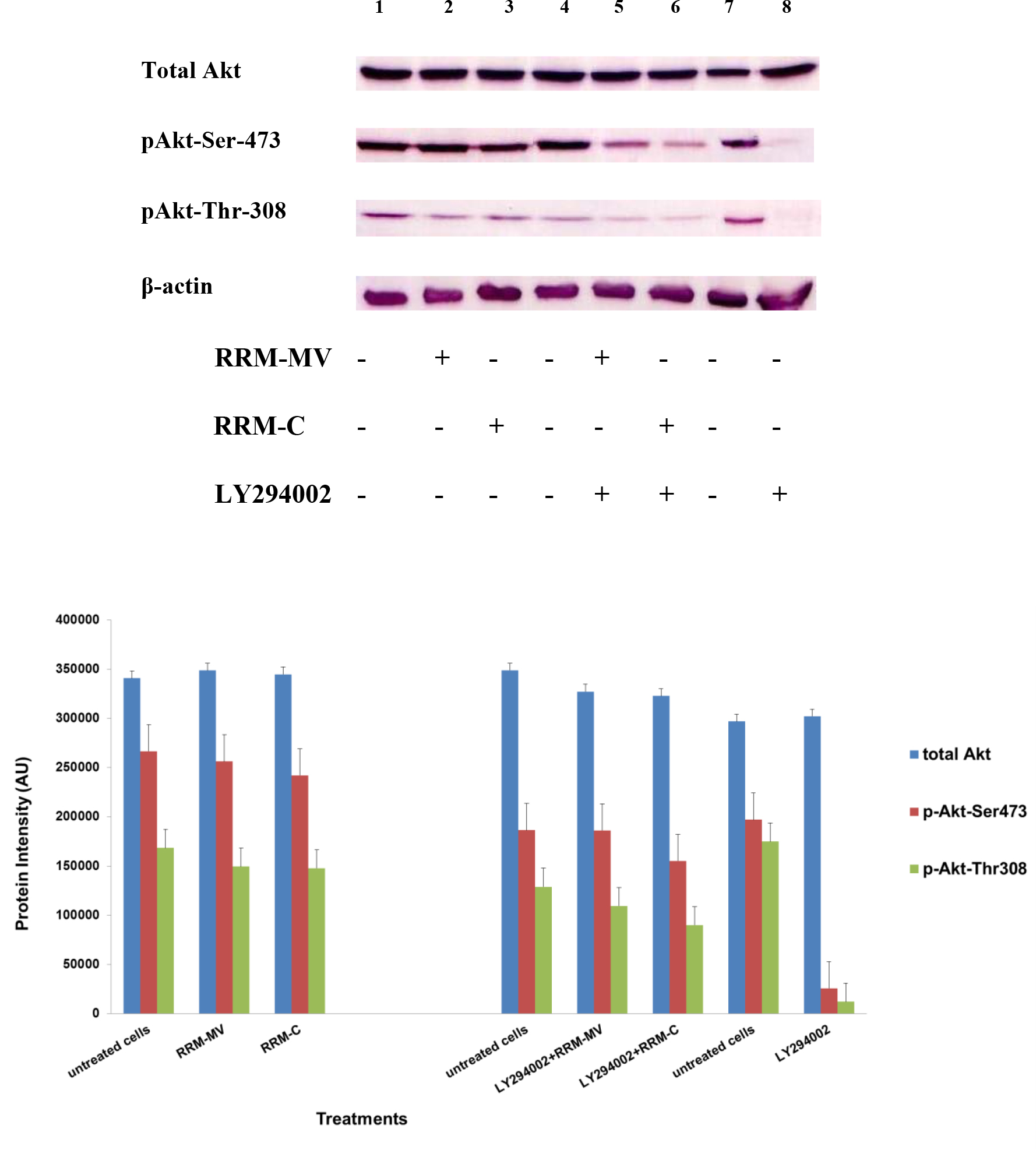
Figure 3. Cellular expression of p-Akt and total Akt in mouse melanoma cells after treatment with the RRM-designed peptides. The B16F0 cell line was treated with 800 ng/mL RRM-MV or RRM-C in the presence or absence of 50 µM LY294002 or treated with 50 µM LY294002 alone for 1 h. The levels of p-Akt expression in the cells were detected by immunoblotting using p-Akt (Ser-473 and Thr-308) and total Akt antibodies. β-actin was used as a loading control. The results of Western blots show that the levels of p-Akt and total Akt in mouse skin cells were not affected by RRM-MV treatment. Inhibition of Akt activity and then treatment with RRM-MV also did not affect expression of p-Akt protein in tested cells. Densitometry of protein spot signals were detected and quantified with the Quantity One Analysis 4.3.0 software. The experiments were performed at least three times.
In normal skin cells: The levels of total Akt protein and p-Akt in the normal non-cancerous HDF cell line were detected after treatment with RRM-MV or RRM-C at the concentration of 400 ng/mL (Figure 4). There were no noticeable changes in the expression of total Akt and p-Akt following RRM-MV treatment when compared to RRM-C treated and untreated cells (Figure 3, lane 2 and 3, respectively). This result suggests that RRM-MV treatment in normal human skin cells did not have any effect on the Akt pathway. As the RRM-MV treatment had no cytotoxic effect on HDF cells [44], no change in the Akt expression levels could be attributed to the finding that RRM- MV has not shown to cause any cytotoxic effects on normal skin cells including HDF.
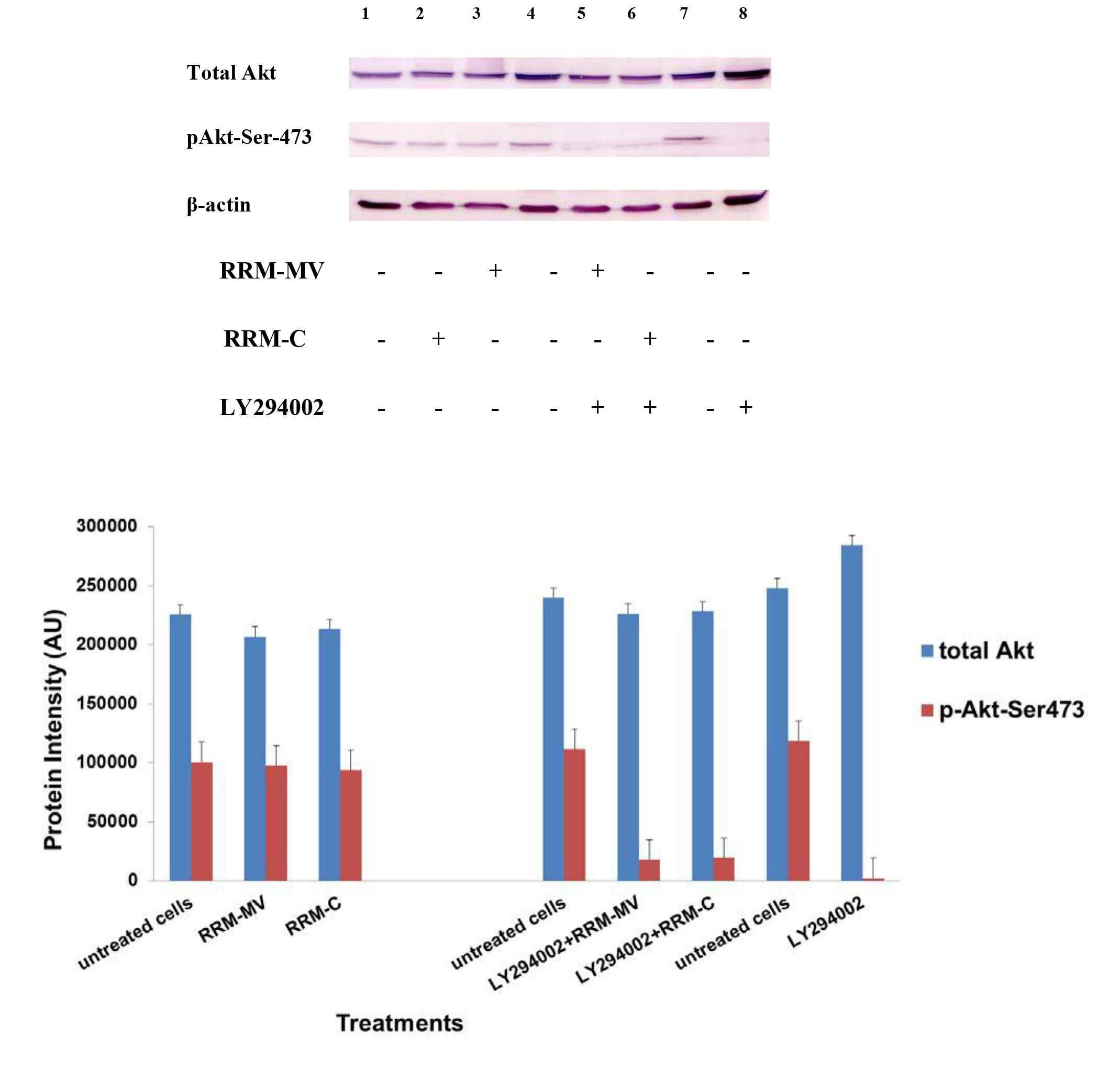
Figure 4. Cellular expression of p-Akt and total Akt in normal human dermal fibroblast cell line treated with the RRM designed peptides. The HDF cell line was treated with 400 ng/mL RRM-MV or RRM-C in the presence or absence of 100 µM LY294002, or treated with 100 µM LY294002 alone for 2 h. Cell lysates were prepared and immunoblotted using p-Akt (Ser-473) and total Akt antibodies. β-actin was used as a loading control. The figure shows RRM-MV treatment did not affect the expression of p-Akt or total Akt in normal human skin cells. Inhibition of Akt activity and then treatment with RRM-MV also had no effect on the expression of p-Akt protein in tested cells. Densitometry of protein spot signals were detected and quantified with the Quantity One Analysis 4.3.0 software. The blots are derived from multiple immunoblotting experiments.
The influence of PI3K Akt inhibitors on Akt activity
Furthermore, to confirm that Akt phosphorylation can be blocked by the PI3K/Akt inhibitor (LY294002) in the previously tested cell lines, cancer and normal skin cells were treated with LY294002 in the presence or absence of the RRM peptides (Figures 1-4). The results showed that incubation of MM96L cell line with 100 µM LY294002 for 1 h blocked Akt phosphorylation either under conditions of complete growth medium or serum starvation compared to untreated cells (Figure 1A and 1B, lane 8). Similar results were observed in B16F0 cell line after treatment with 50 µM of LY294002 for 1 h (Figure 3, lane 8). Nevertheless, the blocking was incomplete at Ser-473 and Thr-308 in COLO-16 cell line in growth medium with serum (Figure 2A, lane 8), yet a complete blocking in phosphorylation of Akt was observed with serum starvation (Figure 2B, lane 8), suggesting that blocking of Akt activation in carcinoma cells was affected by serum conditions. Yet in the HDF normal cell line, treatment with 50 µM LY294002 for 1 h was not sufficient to block the p-Akt pathway (data not shown), therefore, incubation with 100 µM of LY294002 for 2 h was needed to block the Akt phosphorylation (Figure 4). On the other hand, LY294002 treatment did not appear to significantly affect the total Akt expression levels in all tested cell lines.
The influence of RRM-MV treatment on Akt activity in presence of PI3K/Akt inhibitor
To determine whether PI3K Akt inhibitor would suppress Akt activity in the presence of RRM- MV under complete growth medium or serum starvation conditions, cells were treated with LY294002. The cells were then further incubated with RRM-MV and whole cell lysates were immunoblotted with the specific antibodies. The obtained results are shown below.
In skin cancer cells: The MM96L and COLO-16 cell lines were incubated with 100 µM LY294002 for 1 h and then further incubated with 400 ng/mL RRM-MV for 3 h. The Western blot results showed that Akt becomes activated at Ser-473 after RRM-MV treatment in MM96L cells but no p-Akt band was detected at Thr-308 under complete growth medium conditions (Figure 1A, lane 5). Moreover, similar results were found with cells incubated under serum starvation conditions (Figure 1B, lane 5). When Akt activation was blocked in the COLO-16 cell line and then treated with RRM- MV, p-Akt activation was restored at both Ser-473 and Thr-308 under complete growth medium (Figure 2A, lane 5) and under serum starvation conditions (Figure 2B, lane 5). Similar results were obtained with B16F0 cell line treated with 50 µM LY294002 for 1 h and then further incubated with 800 ng/mL RRM-MV for 3 h (Figure 3, lane 5). However, inhibition of Akt phosphorylation followed by treatment with RRM-MV did not have an effect on the expression of total Akt protein in these skin cancer cell lines. The findings of this experiment suggest that LY294002 was not able to block Akt phosphorylation in the presence of RRM-MV in all tested cancer cell lines; thus, confirming RRM-MV treatment interferes with inhibition of Akt signaling pathway.
In normal skin cells: The HDF cell line was incubated with 100 µM of LY294002 for 2 h and then further incubated with 400 ng/mL RRM-MV for 3 h. The Western blot results showed that RRM-MV treatment did not restore p-Akt activation after inhibition with LY294002 (Figure 3, lane 5). This data demonstrate that RRM-MV does not interfere with the p-Akt pathway in normal cells and was unable to restore Akt phosphorylation after inhibition with LY294002.
The influence of different incubation times on Akt expression in cancer cells after RRM- MV treatment
In our previous studies we concluded that the cytotoxic effects of RRM-MV on cancer cells are time- and dose-dependent [43; 44]. To evaluate if time can influence the Akt phosphorylation pathway after RRM-MV treatment leading to cell death in MM96L and COLO-16, p-Akt expression levels were assessed when cancer cells were incubated with 200 ng/mL of RRM-MV for 15 min, 30 min and 60 min under serum starvation conditions. Western blots results showed that in MM96L cell line, the p-Akt S-473 expression was unaffected after 15 min of treatment (Figure 5A, lane 2), but it at was slightly enhanced at 30 min and 60 min (Figure 5A, lane 4 and 6, respectively). As expected, the total Akt expression did not seem to be affected during different incubation times (Figure 5A). Similarly, RRM-MV treatment in COLO-16 did not affect the p-Akt protein levels (Thr-308 and Ser-473) after 15 min (Figure 5B, lane 2); however, phosphorylated Akt was increased slightly at both Thr-308 and Ser-473 after 30 min and 60 min (Figure 5B, lane 4 and 6) respectively. The intensity of total Akt band was unaffected during times course (Figure 5B). These findings indicate that RRM-MV treatment enhanced Akt phosphorylation in a time-dependent manner in melanoma and carcinoma cells.
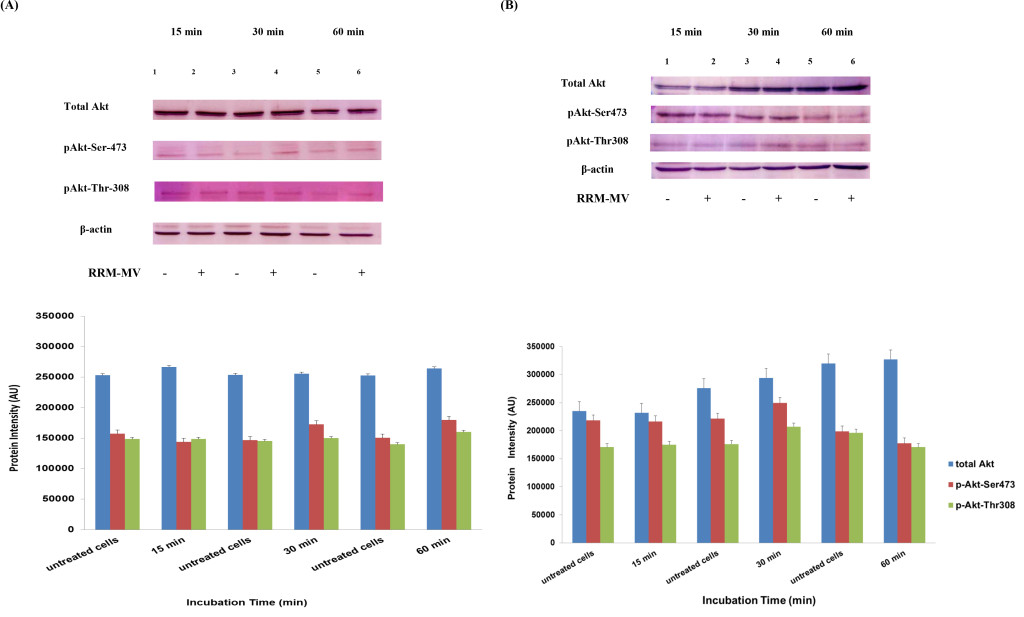
Figure 5. Akt expression levels in melanoma and carcinoma cells during the first hour after RRM-MV treatment. MM96L cell line in (A) and COLO-16 cell line in (B) were treated with 400 ng/mL with RRM-MV for 15 min, 30 min and 60 min. Whole cells extracts were prepared and immunoblotted using p-Akt (Ser-473 and Thr-308) and total Akt antibodies. β-actin was used as a loading control. Western blots show that treatment with RRM-MV induced the levels of p-Akt in a time dependent manner in melanoma and carcinoma cells. Densitometry of protein spot signals were detected and quantified with the Quantity One Analysis 4.3.0 software. The experiments were repeated at least three times.
Discussion
Akt, also known as protein kinase B (PKB), plays a crucial role in the regulation of multiple cellular processes including programmed cell death and it is believed that the regulation of Akt activation is impaired in cancer cells [26]. It was previously shown that M-T5 of myxoma virus binds to p-Akt to regulate Akt signaling in some infected human cancer cells and pharmacological manipulation of AKT phosphorylation, significantly increased MYXV replication in some semipermissive/nonpermissive human cancer cells [26]. It was previously found that RRM-MV, the peptide analogue of the MYXV M-T5 protein, is able to induce apoptotic/ necrotic effects in different skin cancer cell lines but produced no cytotoxic effects on normal non-cancerous cells [43, 44]. In the current study, we investigated the influence of RRM-MV and RRM-C synthetic peptides, on the Akt activation pathway in treated skin cancer cells and in normal skin cells to detect a possible cell death pathway.
One of the findings of the present study is that the expression levels of total Akt protein in all types of tested cells were not affected by the RRM-MV treatment. In addition, a higher level of phosphorylated Akt was detected in RRM-MV treated B16F0 cells, than the p-Akt levels expressed by MM96L and COLO-16 cells at both Ser-473 and Thr-308. This result corroborates with the previous studies [24, 25] indicating that infected MYXV cancer cells expressed measurable phosphorylated Akt levels either high, low or very low levels but some cancer cells did not express any levels of p-Akt. As an example, melanoma (SK-MEL5) cell line expressed total Akt protein but did not express p-Akt (both Ser-473 and Thr-308) [26]. Another example of high levels of Akt is in B16F10 cell line, which correlates with permissiveness to MV [55].
The Western blot results also showed that p-Akt expression was slightly higher at Ser-473 following the RRM-MV treatment, suggesting that RRM-MV may interact with the Ser-473 component of the Akt [56-59]. The RRM-MV treatment did not produce an effect on p-Akt activation in B16F0 cells confirming our previous observations [43].
Our study also showed that different incubation times affected Akt expression in melanoma and carcinoma cells after the RRM-MV treatment. The band’s intensity of p-Akt was unchanged at 15 min of incubation time with RRM-MV while it started to increase after 30 min and 60 min. However, there was no change in the intensity of total Akt band detected during the course of treatment, indicating that Akt phosphorylation was induced by RRM-MV treatment in a time- dependent manner in both melanoma and carcinoma cells. It has been demonstrated that exposure of cells to various chemotherapies and cytotoxic agents leads to changes in the p-Akt expression [60-62] and this also could be the case in RRM-MV treatment. Wang et al. [26] reported that the Akt phosphorylation in MYXV infected permissive cancer cells can be activated by forming a complex between M-T5 and Akt molecule. .
LY294002, which is a potent and specific inhibitor of PI3K that can blocks downstream pathways of PI3K including Akt activation [63-65], blocked p-Akt in tested melanoma and carcinoma cells but it was less effective in normal human skin cells as higher doses and longer incubation time were needed to induce a partial blockage. Also LY294002 was unable to block the total Akt protein levels in all tested cell lines.
We also found that LY294002 did not suppress Akt activity in the presence of RRM-MV in melanoma and carcinoma cells. This result suggests that PI3K kinase inhibitor is unable to block Akt activation following RRM-MV treatment; thus, it is suggested that the RRM-MV has a unique capacity to modulate Akt activation either directly at the Akt level or down-stream the inhibitor binding site in the PI3K pathway [66]. This finding indicated that RRM-MV is able to specifically interact with Akt and that this (RRM-MV-Akt) interaction is required for enhancement of the Akt activation. The findings are in agreement with previous studies by Dhawan et al., [67] and Wang et al., [26] who reported that MYXV infection leads to an increased Akt activation in the presence of LY294002. Hence, it can be concluded that RRM- MV treatment could activate Akt by the phosphorylation of the Ser-473 site. Further work will focus on binding of RRM-MV with tumor specific biomarkers to achieve targeted delivery.
List of abbreviations: B16F0, mouse skin melanoma cell line; COLO-16, human squamous cell carcinoma cell line; HDF, normal human dermal fibroblast cell line; MM96L, human malignant melanoma cell line; MYXV, myxoma virus.
Authors’ contribution: NMA performed experiments and wrote the first draft of the manuscript. TSI and EP designed the experiments and revised the manuscript.
Conflict Of Interest: The authors declare that they have no conflict of interest.
References
- Douek M, Taylor I (2006) Good practice and quality assurance in surgical oncology. Lancet Oncol 4: 626–630.
- Bell JC, Garson KA, Lichty BD, Stojdl DF (2002) Oncolytic viruses: programmable tumour hunters. Curr Gene Ther 2: 243–254.
- Mullen JT, Tanabe KK (2002) Viral oncolysis. Oncologist 7: 106–119. [crossref]
- Yu DC, Working P, Ando D: Selectively replicating oncolytic adenoviruses as cancer therapeutics. Curr Opin Mol Ther 4: 435–443.
- Parato KA, Senger D, Forsyth PAJ, Bell JC (2005) Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer 5: 965–976.
- Fenner F (2000) Adventures with poxviruses of vertebrates. FEMS Microbiol Rev 24: 123–133. [crossref]
- Kerr P, McFadden G: Immune responses to myxoma virus. Viral Immunol 15: 229–246.
- Liu J, Wennier S, Moussatche N, Reinhard M, Condit R, McFadden G (2012) Myxoma Virus M064 Is a Novel Member of the Poxvirus C7L Superfamily of Host Range Factors That Controls the Kinetics of Myxomatosis in European Rabbits. J Virol 86: 5371–5375.
- Stanford MM, Barrett JW, Nazarian SH, Werden S, McFadden G (2007) Oncolytic virotherapy synergism with signaling inhibitors: Rapamycin increases myxoma virus tropism for human tumor cells. J Virol 81: 1251–1260.
- Andrewes Ch, Harisijades S (1955) Propagation of myxoma virus in one-day old mice. Br J Exp Pathol 36: 18–21. [crossref]
- Jackson EW, Dorn CR, Saito JK, McKercher DG (1966) Absence of serological evidence of myxoma virus infection in humans exposed during an outbreak of myxomatosis. Nature 211: 313–314.
- McCabe VJ, Tarpey I, Spibey N (2002) Vaccination of cats with an attenuated recombinant myxoma virus expressing feline calicivirus capsid protein. Vaccine 20: 2454–2462.
- McFadden G (2005) Poxvirus tropism. Nat Rev Microbiol 3: 201–213. [crossref]
- Stanford MM, Shaban M, Barrett JW, Werden SJ, Gilbert PA, et al. (2010) Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther 16: 52–59.
- Lun X, Yang W, Alain T, Shi Z-Q, Muzik H, et al. (2005) Myxoma Virus Is a Novel Oncolytic Virus with Significant Antitumor Activity against Experimental Human Gliomas. Cancer Res 65: 9982–9990.
- Correa RJ, Komar M, Tong JG, Sivapragasam M, Rahman MM, et al. (2012) Myxoma virus-mediated oncolysis of ascites-derived human ovarian cancer cells and spheroids is impacted by differential AKT activity. Gynecol Oncol 125: 441–450.
- Wennier ST, Liu J, Li S, Rahman MM, Mona M, et al. (2012) Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther 20: 759–768. [crossref]
- Hyun T, Yam A, Pece S, Xie X, Zhang J, et al. (2015) Loss of PTEN expression leading to high Akt activation in human multiple myelomas. Blood 96: 3560–3568.
- Testa JR, Bellacosa A (2001) AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A 98: 10983–10985. [crossref]
- Kim CS, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng SY, Ringel MD: AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology 146: 4456–4463.
- Li L, Ittmann MM, Ayala G, Tsai MJ, Amato RJ, Wheeler TM, et al. (2005) The emerging role of the PI3-K-Akt pathway in prostate cancer progression. Prostate Cancer Prostatic Dis 2005, 8(2): 108–118.
- Nicholson KM, Anderson NG (2002) The protein kinase B/Akt signaling pathway in human malignancy. Cell Signal 14: 381–395.
- Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH, Rodrigo JP, et al. (2005) Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer 114: 242–248.
- Shi Y, Gera J, Hu L, Hsu JH, Bookstein R, Li W, Lichtenstein A (2002) Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res 62: 5027–5034.
- Staal FJ, van der Luijt RB, Baert MR, van Drunen J, van Bakel H, et al. (2002) A novel germline mutation of PTEN associated with brain tumours of multiple lineages. Br J Cancer 86: 1586–1591.
- Wang G, Barrett JW, Stanford M, Werden SJ, Johnston JB, et al. (2006) Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc Natl Acad Sci U S A 103: 4640–4645.
- Kim M, Williamson CT, Prudhomme J, Bebb DG, Riabowol K, Lee PWK, Lees-Miller SP, Mori Y, Rahman MM, McFadden G et al. (2010) The viral tropism of two distinct oncolytic viruses, reovirus and myxoma virus, is modulated by cellular tumor suppressor gene status. Oncogene 29: 3990–3996.
- Sypula J, Wang F, Ma Y, Bell JC, McFadden G (2004) Myxoma virus tropism in human tumor cells. Gene Ther Mol Biol 8: 108–114.
- Mossman K, Lee SF, Barry M, Boshkov L, McFadden G (1996) Disruption of M-T5, a novel myxoma virus gene member of poxvirus host range superfamily, results in dramatic attenuation of myxomatosis in infected European rabbits. J Virol 70: 4394–4410.
- Johnston JB, Wang G, Barrett JW, Nazarian SH, Colwill K, et al. (2005) Myxoma virus M-T5 protects infected cells from the stress of cell cycle arrest through its interaction with host cell cullin-1. J Virol 79: 10750–10763.
- Wang G, Barrett JW, Nazarian SH, Everett H, Gao X, et al. (2004) Myxoma virus M11L prevents apoptosis through constitutive interaction with Bak. J Virol 78: 7097–7111.
- Harlan JE, Yoon HS, Hajduk PJ, Fesik SW (1995) Structural characterization of the interaction between a pleckstrin homology domain and phosphatidylinositol 4,5- bisphosphate. Biochemistry 34: 9859–9864.
- Haas-Kogan D, Shalev N, Wong M, Mills G, Yount G, Stokoe D: Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr Biol 8: 1195–1198.
- Dahia PLM, Aguiar RCT, Alberta J, Kum JB, Caron S, et al: (1999) PTEN Is Inversely Correlated With the Cell Survival Factor Akt/PKB and Is Inactivated Via Multiple Mechanisms in Haematological Malignancies. Hum Mol Genet 8: 185–193.
- Amornphimoltham P, Sriuranpong V, Patel V, Benavides F, Conti CJ, et al. (2004) Persistent activation of the Akt pathway in head and neck squamous cell carcinoma: a potential target for UCN-01. Clin Cancer Res 10(12 Pt 1): 4029–4037.
- Aghi M, Martuza RL (2005) Oncolytic viral therapies – the clinical experience. Oncogene 24: 7802–7816. [crossref]
- Chen YL, Law PY, Loh HH (2005) Inhibition of PI3K/Akt signaling: an emerging paradigm for targeted cancer therapy. Curr Med Chem Anticancer Agents 5: 575–589.
- Chow LM, Baker SJ (2006) PTEN function in normal and neoplastic growth. Cancer Lett 241: 184–196. [crossref]
- Hafner C, Houben R, Baeurle A, Ritter C, Schrama D,et al. (2012) Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PLoS One 7: e31255.
- Stiles B, Gilman V, Khanzenzon N, Lesche R, Li A, Qiao R, Liu X, Wu H (2002) Essential role of AKT-1/protein kinase B alpha in PTEN-controlled tumorigenesis. Mol Cell Biol 22: 3842–3851.
- Lawlor MA, Alessi DR (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci 114: 2903–2910. [crossref]
- Cosic I (1997) The Resonant Recognition Model of Macromolecular Bioactivity. Basel, Switzerland: Birkhauser Verlag.
- Istivan TS, Pirogova E, Gan E, Almansour NM, Coloe PJ, Cosic I (2011) Biological effects of a de novo designed myxoma virus peptide analogue: evaluation of cytotoxicity on tumor cells. Plos One 6(9): e24809.
- Almansour NM, Pirogova E, Coloe PJ, Cosic I, Istivan TS (2012) A bioactive peptide analogue for myxoma virus protein with a targeted cytotoxicity for human skin cancer in vitro. J Biomed Sci 19: 65.
- Pirogova E, Vojisavljevic V, Istivan T, Coloe P, Cosic I (2010) Review study: influence of electromagnetic radiation on enzyme activity and effects of synthetic peptides on cell transformation. Med Data Rev 2: 317–324.
- Pirogova E, Istivan T, Gan E, Cosic I (2011) Advances in methods for therapeutic peptide discovery, design and development. Curr pharm biotechnol 12: 1117–1127.
- Pirogova E, Istivan T, Gan E, Coloe P, Cosic I, Dössel O, Schlegel WC (2009) Computationally Designed Interleukin-Like Peptide as a Candidate for Cancer Treatment World Congress on Medical Physics and Biomedical Engineering, September 7 – 12: 1973–1976.
- Almansour NM, Pirogova E, Coloe PJ, Cosic I, Istivan TS (2012) Investigation of cytotoxicity of negative control peptides versus bioactive peptides on skin cancer and normal cells: a comparative study. Future med chem 4: 1553–1565.
- Cosic I, Pirogova E (2007) Bioactive peptide design using the Resonant Recognition Model. Nonlinear Biomed Phys 1: 7.
- Pirogova E., Istivan T. Toward Development of Novel Peptide-Based Cancer Therapeutics: Computational Design and Experimental Evaluation. In: Wang X. (eds) Bioinformatics of Human Proteomics. Translational Bioinformatics, 2013, vol 3, Springer, Dordrecht, ISBN 978-94-007-5810-0, pp 103–126.
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72: 48–254.
- Bidwell GL, 3rd, Raucher D (2009) Therapeutic peptides for cancer therapy. Part I – peptide inhibitors of signal transduction cascades. Expert Opin Drug Deliv 6: 1033–1047.
- Raucher D, Moktan S, Massodi I, Bidwell GL (2015) 3rd: Therapeutic peptides for cancer therapy. Part II – cell cycle inhibitory peptides and apoptosis-inducing peptides. Expert Opin Drug Deliv 6: 1049–1064.
- Park YJ, Chang LC, Liang JF, Moon C, Chung CP, Yang VC (2010) Nontoxic membrane translocation peptide from protamine, low molecular weight protamine (LMWP), for enhanced intracellular protein delivery: in vitro and in vivo study. FASEB J 19: 1555–1557.
- Stanford MM, McFadden G (2007) Myxoma virus and oncolytic virotherapy: A new biologic weapon in the war against cancer. Expert Opini Biol Ther 7: 1415– 1425.
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. (1997) Greenberg ME: Akt Phosphorylation of BAD Couples Survival Signals to the Cell-Intrinsic Death Machinery. Cell 91: 231–241.
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, et al. (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661–665.
- Downward J (1998) Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol 10: 262–267.
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, et al. (1999) Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 96: 857–868.
- Tang D, Okada H, Ruland J, Liu L, Stambolic V, et al. (2001) Akt is activated in response to an apoptotic signal. J Biol Chem 276: 30461–30466. [crossref]
- Winograd-Katz SE, Levitzki A (2006) Cisplatin induces PKB//Akt activation and p38MAPK phosphorylation of the EGF receptor. Oncogene 25: 7381–7390.
- Jetzt A, Howe JA, Horn MT, Maxwell E, Yin Z, Johnson D, Kumar CC (2003) Adenoviral- Mediated Expression of a Kinase-Dead Mutant of Akt Induces Apoptosis Selectively in Tumor Cells and Suppresses Tumor Growth in Mice. Cancer Res 63: 6697–6706.
- King WG, Mattaliano MD, Chan TO, Tsichlis PN, Brugge JS (1997) Phosphatidylinositol 3- kinase is required for integlrin-stimulated AKT and Raf-1/mitogen-activated protein kinase pathway activation. Mol Cell Biol 17: 4406–4418.
- Wennström S, Downward J (1999) Role of Phosphoinositide 3-Kinase in Activation of Ras and Mitogen-Activated Protein Kinase by Epidermal Growth Factor. Mol Cell Biol 19: 4279–4288.
- Werden SJ, McFadden G (2002) Pharmacological manipulation of the akt signaling pathway regulates myxoma virus replication and tropism in human cancer cells. J Virol 84: 3287–3302.
- Soares JAP, Leite FGG, Andrade LG, Torres AA, De Sousa LP, Barcelos LS, Teixeira MM, Ferreira PCP, Kroon EG, Souto-Padrón T et al: (2009) Activation of the PI3K/Akt Pathway Early during Vaccinia and Cowpox Virus Infections Is Required for both Host Survival and Viral Replication. J Virol 83: 6883–6899.
- Dhawan P, Singh AB, Ellis DL, Richmond A (2002) Constitutive Activation of Akt/Protein Kinase B in Melanoma Leads to Up-Regulation of Nuclear Factor-KB and Tumor Progression. Cancer Res 62: 7335–7342.